
Introduction
Lung adenocarcinoma (LA) is the most common cause of cancer-related death worldwide [1–8]. Despite the advances over the last decade in new targeted therapies, cancer genetics, diagnostics, staging, and surgical techniques as well as new chemotherapy and radiotherapy protocols, the death rate from LA remains high [3, 4, 9–11]. A number of strategies have been employed. Early LA is asymptomatic, and the vast majority of patients are diagnosed at an advanced stage. LA is an umbrella term, under which fall multiple histologic variants that are mainly composed of an array of heterogeneous molecular subtypes, most of them characterised by a single oncogenic alteration [1, 3, 8]. From a clinical and pathological standpoint, the vast majority of LAs are diagnosed using a single adenocarcinoma marker (e.g. TTF-1 or mucin) and are grouped histologically according to tissue morphology as epithelial lung tumours, which are further subclassified into: lipidic adenocarcinoma; acinar adenocarcinoma; papillary adenocarcinoma; micropapillary adenocarcinoma; solid adenocarcinoma; invasive mucinous adenocarcinoma, with its variants mixed invasive mucinous and non-mucinous adenocarcinoma; colloid adenocarcinoma; foetal adenocarcinoma; enteric adenocarcinoma; minimally invasive adenocarcinoma, with two types: mucinous and non-mucinous; and preinvasive lesions: atypical adenomatous hyperplasia and adenocarcinoma in situ, which can be mucinous or non-mucinous [4, 12–14].
LAs arise mostly from distal airways, commonly with glandular histology displaying biomarkers that are consistent with an origin in the distal lung, including thyroid transcription factor 1 (NKX2-1) and keratin [3, 13, 15–17].
Unfortunately, the clinical, radiological, molecular, and pathological spectrum is widely divergent in terms of overall survival and prognosis due to histological heterogeneity of lung adenocarcinomas [4]. Accordingly, there is a critical need to elucidate the molecular mechanisms associated with the tumoural spreading process and therapeutic resistance of this serious pathology.
The tumour microenvironment is composed of several cytokines, one of which is transforming growth factor β1 (TGF-β1), which modulates and mediates the expression of epithelial-mesenchymal transition (EMT), correlated with invasive growth in LAs, and exhibits its pleiotropic effects through binding to transmembrane receptors TβR-1 (also termed activin receptor-like kinases [ALKs]) and TβR-2. TGF-β is expressed on several malignancies, including LAs [9, 18–25].
TGF-βsignalling promotes the migration of tumour cells to metastasis sites. Humans express three highly potent homologous cytokine isoforms of TGF-β consisting of TGF-β 1, 2, and 3. In LA carcinogenesis TGF-β1 plays an important role in regulating cellular functions such as cell proliferation, differentiation, tumour suppression, epithelial inflammation, cell motility, apoptosis, adhesion, invasion, and immune response [26–29]. Functionally, TGF-β1 cytokines exert pro-inflammatory effects in epithelial tumourigenesis. Initially, TGF-β1 acts as a tumour suppressor. At later stages, as the tumour grows, TGF-β1 is produced by both tumour and stromal cells as a natural response to hypoxic and inflammation [30].
In this review, the current role of contextual signal TGF-β1 inducer of epithelial mesenchymal transition in patients with lung adenocarcinoma, and an overview of our current mechanistic understanding of the TGF-β1-related pathways in brain metastasis progression-inhibitors that could be used for clinical treatment, as well as an examination of the models used to study these processes, will be briefly discussed. Finally, current progress in the therapeutic approaches targeting TGF-β1 will be summarize.
TGF-β1 signalling pathway plays a critical role in EMT expression in lung adenocarcinomas
Several studies have reported that TGF-β1 is involved in the tumorigenesis of lung adenocarcinoma cells. The TGF-β superfamily, first coined as sarcoma growth factor by de Larco and Todaro in 1978 [5, 31], seems to regulate the transdifferentiation process of EMT. Importantly, TGF-β1 protein has been shown to play a critical role in the development of EMT [1, 15, 18, 22, 31–62]. However, despite some excellent published work, the role of TGF-β1 on cellular proliferation, invasiveness, immune response, angiogenesis, differentiation, oxidative stress, and metastasis pathophysiology remains to be fully elucidated. EMT is a well-studied biologic phenotypic switching process by which epithelial cells lose their characteristic signals and cell adhesion properties converting into motile mesenchymal cells [9, 19, 22, 47]. Three classes of EMT events have been identified. Type 3 EMTs occur in tumour cells [9, 20].
On the other hand, EMT play a pivotal role in the context; it was shown that TGF-β1 coordinates and EMT plays an important role in control of growth, survival, and metastasis [39, 59]. TGF/EMT signalling, which is mainly mediated by RAS-MAPK, a driver and critical downstream pathway of human carcinomas, and PI3K-AKT/HIF-1a pathways affect gene expression and cell cycle progression through the binding of transcription factors [19, 45] such as the E26 transformation-specific (ETS) family, regulating EMT by enhancing the expression of EMT-transcription factors including ZEB1, Snail, VEGF, etc. [19, 42, 63–65]. Cytoplasmic signalling cascades mediated by PI3K-AKT and the GTPase RAC1 or cell division control protein 42 (CDC42) is found to be altered in several stages of tumour progression. MMP-3 increases the level of RAC1b, which gives rise to cellular ROS, leading to EMT and oxidative damage to DNA, modulating cell survival, and eliciting actin and microtubule cytoskeletal changes, regulation of gene expression, cell polarity, and cell migration [10, 17].
The TGF-β1 signalling is regarded as an initial, complex, and yet crucial cytokine to induce EMT during cancer progression and metastasis in a Smad2/Smad3 complex-dependent C-terminal phosphorylated downstream manner via ALK-5 receptors, and also dependent on the protein-serine/threonine kinases that participate in the Ras-Raf-MEK5-ERK5 signal transduction cascade and nuclear translocation [1, 15, 18, 22, 25, 29, 31, 33, 45, 47–49, 57, 65–72], with positive enhancement of T-cell and cytokine production [73]. Subsequently this Smad2/Smad3 bond forms a special hetero-oligomeric link with common mediator Smad4 giving way to a SMAD2/3/4 translocating complex into the nucleus, where it binds to transcription factors Snail/Slug, Twist, and Zeb, subsequently modulating the transcription of target genes that regulate cell-cell and ECM interactions, thus providing a favourable microenvironment for tumour cell spread [18, 47–49, 71, 72, 74]. TGF-β1 acts as a pleiotropic factor and cytokine, promoting cell proliferation, survival, motility, scattering, differentiation, and morphogenesis. Physiologically, EMT is responsible for the cell-scattering phenotype. This process involves the disruption of cadherin-based cell-cell contacts and subsequent migration of the dissolved cells into the circulation [75]. High TGF-β signalling promoting EMT invasiveness and metastatic growth is achieved by ligand binding to TGF-β receptor II, which associates with the type I receptor activin receptor-like kinase-5 (ALK-5) through the subsequent phosphorylation of Smad2 and Smad3, which is constitutively regulated by the cytoplasmic protein profilin-2 (PFN2) [23, 75]. However, a more recent in vivo liver and lung metastasis mouse model study investigating whether PFN2 served specific roles in the progression of human colorectal cancer demonstrated, by reverse transcription-quantitative polymerase chain reaction and western blotting techniques, that PFN2 expression was reduced in metastatic CRC [69].
Molecularly, during embryogenesis TGF-β cytokine leads several biological processes such as cell cycle and apoptosis, EMT, and extracellular matrix (ECM) regulation. Recent studies investigating molecular mechanisms and epigenetic alterations of carcinogenesis have indicated that Smad6 antagonises Smad2-4 complex activation by competing with Smad2-4 binding to the activated TGF-β1 receptor [76] and that bone morphogenetic protein (BMP)-mediated SMAD signalling requires an arginine methylation step for the removal of an otherwise signalling-inhibiting SMAD6 protein ahead of transphosphorylation of the TGF-β/BMP receptor kinases [3, 13, 76]. Furthermore, emerging evidence supports the theory that SMAD6 inhibits BMP signalling by interacting with homeobox C8 in the nucleus, and Smad6 antagonises TGF-β1 in the cytoplasm through the formation of a stable complex with TGF-βR1. In contrast, SMAD7 (mothers against decapentaplegic homolog 7) inhibits TGF-β, activin, and BMP signalling. Smad7 interacts with TGF-βR1 to inhibit R-smad activation and is commonly found in patients with chronic inflammatory conditions of the colon and also enhances the invasiveness of non-small-cell lung cancer cells. Recent studies have shown that Smad7 can be promoted and targeted by micro-RNAs (miR), such as miR-106b-25 and miR-21-5p cluster, during EMT in breast cancer development and NSCLC proliferation, respectively [77, 78].
Although the importance of epigenetic mechanisms regulating TGF-β1 has been recognised, several groups have reported genome-wide analyses of the binding patterns of TGF-β1 receptor-regulated Smads in various cancer cell lines and embryonic stem cell-derived cells. Various Smad-binding profiles have been revealed in different cell types, indicating that the cell environment is of paramount importance for the response to signalling [3–5, 11, 23, 36, 43, 44, 48, 67, 77, 79–84].
The combination of in vitro cell-based or in vivo mouse-based models has been widely used to interrogate the dynamic behaviour of TGF-β1 signalling networks. However, the majority of models have been created by directly inactivating the expression of negative feedback pathway regulators, such as the E3 ubiquitin ligase SMURF2 and SMAD7 of the TGF-β superfamily pathway [17, 32–36, 77, 78, 85–87]. Considering its inhibitory roles in the TGF-β1 receptor, inhibitory SMAD proteins may play a critical role by inhibiting the development of endothelial cells. Furthermore, distinctive canonical (Smad-dependent) and non-canonical (Smad-independent) (Fig. 1) TGF-β superfamily member inhibitors have been identified using human and animal models [60] such as GDF15 (identified as a critical downstream mediator of CDP138, and described as a CDK5 binding partner that regulates cell proliferation and migration), which attenuates the TGF-β/Smadsignalling pathway, leading to impaired radio-resistance and metastasis in lung cancer [10]; CK2 (formally named casein kinase 2) inhibitor CX-4945 affects the TGF-β1-induced cadherin switch and the activation of the smad, non-smad, and focal adhesion signalling pathways, avoiding the migration and invasion of A549 human lung epithelial adenocarcinoma cells [61]; Bcl-xL, an anti-apoptotic and probably antioxidant protein overexpressed in LAs, has been demonstrated to neutralise the pro-apoptotic functions of Bcl-2-associated X protein (Bax) and Bcl-2-associated death promoter (Bad) through mitochondrial membrane permeability integrity, which mechanistically preclude pro-caspace-9 activation and cytochrome c release [50, 88, 89], among other factors. However, although these inhibitor pathways are attractive therapeutic targets in LAs [90], mounting evidence indicates that Smad/ TGF-β/EMT axis suppression, like most complex biological pathways, leads to compensatory over-activation of upstream pathways, resulting in sustained inhibition of the anti-angiogenic, anti-proliferative, and anti-metastatic effects of the recognised inhibitors. In order to examine and to understand the mechanism of the therapeutic potential of targeting TGF-β, it is necessary to elucidate the full spectrum of feedback loops that are unleashed by suppression of the smad/ TGF-β/EMT axis [60, 81, 91].
Fig. 1
Canonical (Smad-dependent) and non-canonical (Smad-independent) pathways. An oversimplified and modified (from [30]) scheme depicting the TGF-β pathway as well as a model of cerebral metastasis from lung adenocarcinoma. A) Disaggregation of cells from primary tumour mass to a particular metastatic niche is mediated mainly through integrins and proteases expressed at the surface of cancer cells, vasculature, and stromal cells [48]. In summary, cells [*] break through the vascular basement membrane via the expression of cellular matrix metalloproteinase activity – MMP2 and MMP9. Many angiogenic factors secreted by both tumour and stromal cells, such as VEGF and platelet-derived growth factor β (PDGF-β), which are transcriptionally directly driven by hypoxia-inducible factor 1 or 2 (HIF1–HIF2). B) Lung adenocarcinoma is commonly defined as a slow-growing cancer that arises mostly from distal airways, typically with glandular histology displaying biomarkers that are consistent with an origin in the distal lung, including thyroid transcription factor 1 (NKX2-1) and keratin (7) with complex early diagnosis because it usually involves the periphery of the lung and has scarce symptoms, promoting early metastasis. C) The TGF-β1 signalling is regarded as an initial, complex, and yet crucial cytokine to induce EMT during cancer progression and metastasis (20) in a Smad2/Smad3 complex-dependent C-terminal phosphorylated downstream manner via ALK-5 receptors, and also dependent on the protein-serine/threonine kinases that participate in the Ras-Raf-MEK5-ERK5 signal transduction cascade and nuclear translocation
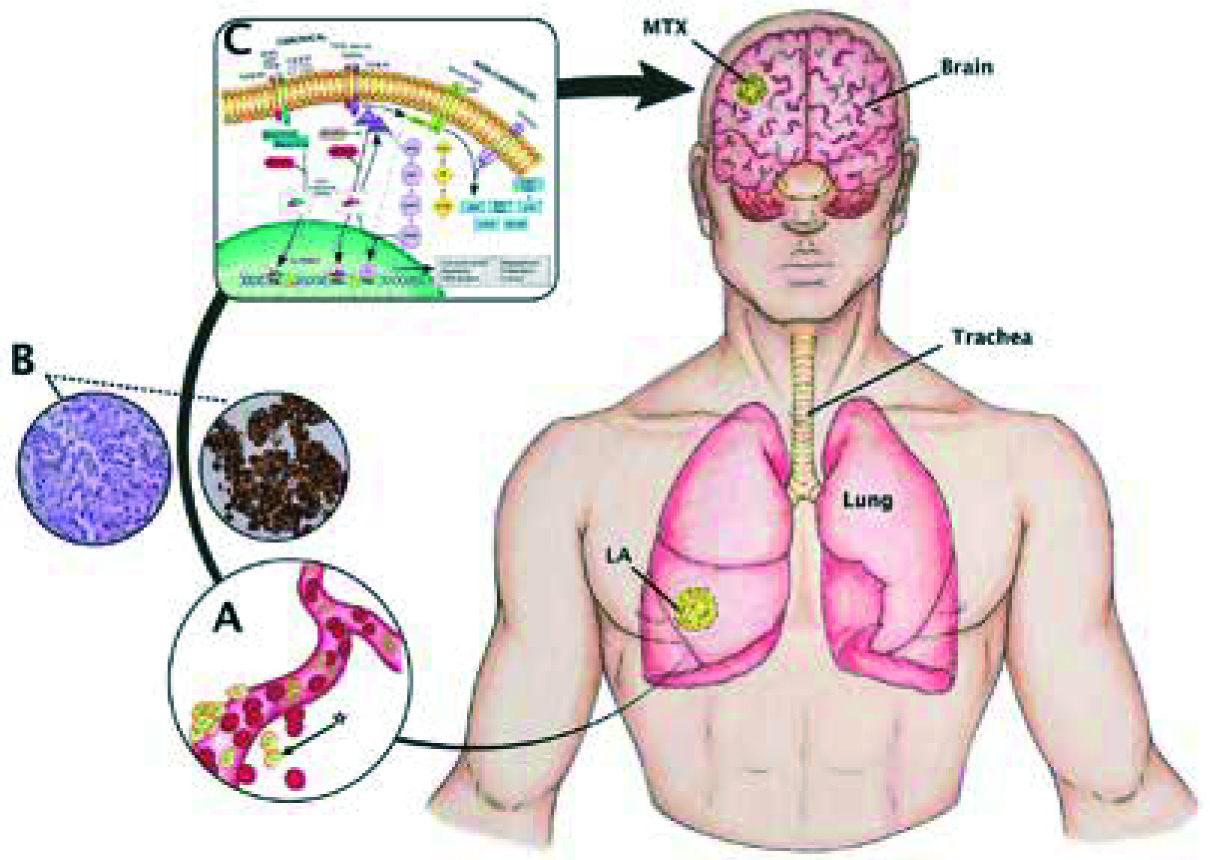
Carcinogenesis and evolution. Brain metastases from lung adenocarcinoma
Metastatic tumours are the most common neoplasms encountered in the brain. Brain metastases most commonly arise from lung, breast, genitourinary tract, and melanoma [41, 42, 55, 92]. Once brain metastases have developed, survival is commonly short, sometimes secondary by a scarce follow-up.
Brain metastasis is a multistep process that involves multiple interactions between the tumour cells and the matrix proteins, with the microenvironment playing a key factor, characterised by abnormal changes at the cellular, genetic, and epigenetic levels. Initiation and development of lung adenocarcinoma is partially attributed to the dysregulation and aberrant expression of chromosomes and proto-oncogenes, ranging from simple structural rearrangements and gene amplifications to losses or gains of entire chromosomes, which lead to cell growth, metastasis, and other tumour progressions. Several proto-oncogenes have been reported in LAs, such as ALK, proto-oncogene tyrosine-protein kinase (ROS1), c-Met, and RET proto-oncogene [58, 67, 70, 93]. Furthermore, it has been established that many miRNAs are down-regulated or up-regulated in certain types of cancers. MicroRNAs (miRNAs) are short endogenous non-coding RNAs implicated in the regulation of gene expression of many relevant physiological processes, including cell proliferation, cell death, and stress responses [35], and thus key drivers of pathological control. It is estimated that up to 30–50% of all human protein-coding genes are regulated by miRNAs [94].
Mounting evidence suggests that miRNAs are deregulated in various human cancers, including LAs, by regulating expression of multiple target genes [52]. They function as oncogenes or tumour suppressor genes. In the context of cancer, a recent study using TGF-β1 in different concentrations to induce EMT in lung cancer A549 cells demonstrated that the lung cancer A549 cells became elongated and the cell-cell junction became loose after EMT [24, 29], resulting in E-cadherin downregulation and the mesenchymal markers vimentin and fibronectin being up-regulated, supporting the theory that EMT has an effect on the expression of miRNAs, and regulating metastasis of lung cancer cells via EMT [34]. To investigate the function of miR-26a in the development of lung cancer, based on recent published data, in order to find prognosis-related miRNA, miR-29c isoform, a member of miR-29 family, aberrantly expressed in different types of human cancers and functionally involved in cell proliferation, cell cycle, apoptosis, and metastasis, has been demonstrated to revert aberrant methylation by targeting DNA methyltransferases 3A and 3B and inhibits cell proliferation, migration, and invasion in cell lines by targeting integrin β1 and matrix metalloproteinase 2 expression [74, 95]. Interestingly enough, miR-29c may target different genes and pathways to inhibit tumour growth and metastasis, implicating miR-29c as a novel promising prognostic biomarker as well as a therapeutic target to provide clinical benefit, through modulating proliferation and migration/invasion of LA cells by VEGFR-2l [29].
Notably, increasing evidence has demonstrated that TGF-β through EMT induces the expression of matrix metalloproteinases on both endothelial cells and tumour cells, which then facilitates the differentiation into stromal stem cells such as pericytes, and is able to create a tissue environment permissive to the metastatic dissemination and colonisation of distant organs [36, 40]. More specifically, cells break through the vascular basement membrane via the expression of cellular matrix metalloproteinase activity – MMP2 and MMP9. Many angiogenic factors secreted by both tumour and stromal cells in the brain, such as VEGF and platelet-derived growth factor β (PDGF-β) are transcriptionally directly driven by hypoxia-inducible factor 1 or 2 (HIF1–HIF2) [19, 29, 73].
Additionally, acidosis, cellular stress, and hypoxia lead to decreased expression of E-cadherin and occludins, resulting in less cell adhesion and entry of the dissolved or circulating tumour cells (CTCs) intro the bloodstream, some of which express the EMT phenotype [20]. E-cadherin is a calcium-dependent cell adhesion glycoprotein abundantly expressed by surface epithelial cells that function as an inhibitory factor for malignant transformation, cell dissociation, and metastasis. Therefore, E-cadherin is a rate-limiting step of invasion and differentiation of cells, downregulated in epithelial cancers, categorised as a marker for EMT. In fact, EMT is well known as a cellular program characterised by loss of an epithelial gene expression signature, such as E-cadherin, and gain of genes that define mesenchymal features, such as vimentin and neural cadherin (N-cadherin) [9, 18, 34, 63, 96, 97].
At epigenetic level, increased Smad2 and Smad3 expression operates in the lung adenocarcinomas and brain metastasis, enhancing TGF-β1 and the production of angiogenic factors such as VEGF [23, 83, 95]. Some investigators have also focused on the role of vascular basement membrane (VBM) in the pathogenesis of brain metastasis [92, 98]. Concordantly, VBM promoted adhesion and invasion of malignant cells. Disaggregation of cells from the primary tumour mass to a particular metastatic niche is mediated mainly through integrins and proteases expressed at the surface of cancer cells, vasculature, stromal cells, etc. [92]. The β1 subunit of integrin is an adhesion molecule involved in cell survival and cancer resistance to radio and chemotherapy. Cooperation between β1 integrin and c-Met regulates tyrosine kinase inhibitor resistance in lung cancer. Blockade of the β1 integrin subunit in tumour cells prevented adhesion to VBM and attenuated metastasis establishment and growth in vivo [29, 34, 38, 41, 58].
Recent insights into the brain tumour microenvironment have begun to uncover the close association between metastatic cells and the blood-brain barrier, by disrupting the endothelium through the vascular basement membrane to gain entry into the circulation and promoting tumour cell dedifferentiation transcriptionally. The VBM serves also as a reservoir for growth factors, such as TGF-β1 and vascular endothelial growth factor (VEGF), which reduce the endothelial barrier function by disrupting the E-cadherin–β-catenin complex and therefore favouring endothelial cell junction opening [26, 84, 99].
Interestingly, bevacizumab is a humanisedMAb targeting VEGF. The inhibition of VEGF signalling via bevacizumab treatment may normalisetumour vasculature, promoting a more effective delivery of chemotherapy agents. A randomised phase III trial (ECOG 4599) combining paclitaxel and carboplatin with or without bevacizumab in patients with advanced LA found a significant improvement in median survival for patients in the bevacizumab group, with a total of 5 of 10 treatment-related deaths occurring as a result of haemoptysis, all in the bevacizumab group [100]. Indeed, the median survival was 12.3 months in the group assigned to chemotherapy plus bevacizumab, as compared with 10.3 months in the chemotherapy-alone group (p = 0.003). In the former study, VEGF levels did not correlate with overall survival. In addition to distant invasion, another characteristic gained by metastatic cells is the adaptive and disorganised formation of new blood vessels with ultrastructural abnormalities from pre-existing vessels possibly mediated by VEGF. Conversely, a recent study found that the treatment with cisplatin/gemcitabine/bevacizumab (PGB) was superior to erlotinib-bevacizumab treatment in patients displaying a mesenchymal phenotype (low E-cadherin or high vimentin), but not in those with an epithelial phenotype (high E-cadherin or low vimentin) [101]. VEGF binds to precursors of endothelial cells via transmembrane receptors of the tyrosine kinase family, flt-1, and FLK-1/KDR, promoting the expansion, migration, and differentiation of vascular networks [23, 95].
In previous research on coculture in vitro experiments by injecting human A375 parental cells into the internal carotid artery of nude mice, astrocytes were found to be involved as critical protectors of the tumour cells from 5-fluorouracil and cisplatin-induced apoptosis in human melanoma cells [102]. Moreover, Chu et al. found that astrocytes produce SDF-1a, IL-3, IL-6, interferon-y, TNF-a, TGF-β, and PDGF, which the tumour cells use to promote survival, growth, and potentially organ-specific metastasis [1, 3]. The essential role of the tumour microenvironment in cancer progression, the paracrine growth factors involved, and the interactions between tumour cells and the brain microenvironment have been elegantly demonstrated by isolation of a brain-specific metastatic variant of the MDA-MB-231 human breast cancer cell line, one of the most metastatic cell lines, through repeated selection in vivo by Bos et al. [38]. In an early study investigating the molecular mechanisms of angiogenesis in experimental brain metastasis, it was found that tumour cells with high rates of metastatic spread to the brain overexpressed significantly higher levels of VEGF activity than tumour cells with low rates of brain metastasis, and also suggesting that VEGF is necessary but not sufficient for the production of brain metastases [103].
Therapeutic potential of TGF-β1 inhibitors in lung adenocarcinoma
Targeted therapies have a response rate of up to 80%, but progression almost always occurs within one to two years. Traditional chemotherapy has a response rate of about 20% with one-year survival rates near to 30%. TGF-β1 is the most abundant isoform in mammals. At present there is a prospective randomised trial exploring the therapeutic options. For example, there are several pharmacokinetic approaches and ongoing trials that interfere with the EMT mechanism and the migration of cancer cells induced by TGF-β1, such as BIX02189.
The downregulation or inhibition of TGF-β1 (the most abundant isoform in mammals) receptors results in cellular resistance to the usual suppressive effects of the TGF-β1 ligand, contributing to cancer development and apoptosis. Existing therapies that target TGF-β1 receptors in a variety of human cancers for potential clinical use are based on antisense molecules (oligonucleotides), monoclonal antibodies against the receptors, soluble TGF-β inhibitors, receptor kinase inhibitors, peptide aptamers, and human vaccines. These therapies were designed to act against specific targets, including Smad proteins and other targets downstream the TGF-β1 signalling pathway. Nonetheless, despite its initial promise, currently many targeted therapies either against ligand, ligand-receptor, or intracellular level of the TGF-β1 on lung adenocarcinoma, which are considered relevant, have been discarded from both in vitro and in vitro research.
Due to brain metastasis from lung adenocarcinoma and its highly complex microenvironment, it is difficult to find a fully comprehensive and effective therapeutic approach. The ability of therapeutic strategies targeting the activating or inhibitory receptors on TGF-β1 to stop or reverse the EMT has been reported in A549 lung cancer cells [15, 104]. In an experimental model on cultured human A549 cells investigating the involvement of ERK1/2 in phosphorylation of Smad3 linker region and EMT induced by TGF-β1, it was found that kaempferol, a common natural flavonoid, acts as a potent antitumour growth agent by reversing TGF-β1-mediated Snail induction and E-cadherin repression by weakening Smad3 binding to Snail promoter [105].
The role of the immune system in cancer progression has been studied for decades. Programmed death-ligand 1 (PD-L1) is a 40kDa type transmembrane protein, a member of the B7-CD28 immunoglobulin superfamily expressed on activated T-cells and B-cells, with an important role in mediating immune evasion in the tumour microenvironment closely related to the EMT process through a negative feedback loop. An outstanding recent study reported that the AKT, ERK, and TAK1 pathways regulated the expression of PD-L1 by mediating transportation of the transcription factor Stat3 and the p65 subunit of NF-kB from the cytoplasm to the nucleus, with such findings determined by western blotting and flow cytometric analyses [63].
Recently, by investigating volatile anaesthetic agents such as sevoflurane on cell viability and chemoresistance to cisplatin on LA A549 cells in an in vitro study, it was found that sevoflurane positively upregulated expression of nuclear Smad3 and TGF-β1 with enhanced chemosensitivity to cisplatin but without effect on migration of A549 cells [44].
As previously mentioned, MEK/ERK5 (mitogen-activated protein kinase/extracellular signal-regulated kinase [ERK]5) signalling pathway, strongly linked to chemoresistance, encoded by MAP2K5, can be inhibited in human A549 lung cancer cells. BIX02189, a pharmacological inhibitor of the MEK5 signalling pathway, has been shown to significantly interfere with the EMT mechanism and in the migration of cancer cells induced by TGF-β1 [27, 56, 64]. Furthermore, a previous study has further shown that cyclooxygenase-2 (COX-2) inhibitors enhance the MEK/Snail1 signalling and lead to metastasis and chemoresistance via EMT induction in NSCLC [106].
Antisense oligonucleotides are a class of molecule that can specifically bind to RNA target molecules in order to manipulate gene expression. Unfortunately, there is no therapy approved by the US Food and Drug Administration for the treatment of LAs, but it may be a feasible approach to minimise levels of TGF-β1 [73].
Fresolimumab, a human neutralising antibody of all mammalian active isoforms of TGF-β, commonly well tolerated, was developed initially in a phase I study to treat patients with advanced melanoma and renal cell carcinoma, with contradictory results [107].
Conclusions
Additional challenges will be faced, shaping the design of clinical trials in cancer biology and pathway targets. In recent years, several researchers have focused on developing TGF-β1 pathway inhibitors, and immense effort has been made to investigate methods for selective eradication of therapeutically resistant cells in lung adenocarcinoma.
The goal of the present review was to give a brief and non-conclusive overview of the most recent data about the implication of TGF-β1 receptors and ligands on lung adenocarcinoma related to the contextual EMT and the multiple therapeutic strategies that have been investigated for improving the effectiveness of TGF-β1 inhibitors. As noted, TGF-β1 is frequently identified as being integral in regulating invasiveness and metastasis in a variety of human cancer types and represents an opportunity to develop therapeutic approaches.






