
Introduction
Silver-Russell syndrome (SRS) is a rare, heterogeneous genetic disease that occurs in both sexes with the same frequency of 1:30,000–1:100,000. The causes of this syndrome include hypomethylation of the 11p15 region of the paternal chromosome, maternal chromosome 7 disomy, or submicroscopic chromosome rearrangements. Molecular testing can confirm the diagnosis only in around 60% of patients, so currently SRS is a clinical diagnosis based on a combination of characteristic features [1]. At present, the Netchine–Harbison clinical scoring system is the main scoring scale for the diagnosis of SRS. It contains 6 criteria [2]:
small for gestational age (SGA),
postnatal growth failure,
relative macrocephaly at birth,
prominent forehead,
body asymmetry,
feeding difficulties and/or low body mass index (BMI).
Clinical diagnosis is considered if a patient scores at least 4 of 6 from these criteria, including both prominent forehead and relative macrocephaly.
Children with SRS suffer from poor appetite, feeding difficulties, and gastrointestinal problems, which additionally can lead to calorie-related length deficiency. On account of their low muscle and liver mass, a disproportionately large brain-for-body size, and feeding difficulties, hypoglycaemia and its neurocognitive consequences are a significant and often irreversible threat [1, 3, 4]. In Poland the treatment of these children with recombinant human growth hormone (rhGH) has been covered since January 2015 by a therapeutic program for short stature children born small for gestational age.
Duchenne muscular dystrophy [DMD] is a genetic entity that occurs only in boys, with a frequency of 1 : 3500 births. The cause of this disease is the mutation of the maternal DMD gene located on the X chromosome, encoding dystrophin. As a result, muscle fibres are easily damaged during contraction, leading to progressive weakness and skeletal muscle degeneration. Usually, the lower external muscles are affected before the upper. Later on, the heart and respiratory muscles are involved. At present, corticosteroids are the standard supportive treatment for DMD, being the only drugs known to slow down the deterioration of muscle strength and motor function. The following 2 corticosteroids are the ones most commonly used in DMD treatment: prednisone (0.75 mg/kg/day) and deflazacort (0.9 mg/kg/day). The age at which loss of independent walking is observed has increased over time. Maintenance of ambulation ensures slowing of the progression of scoliosis. Moreover, boys with DMD are at higher risk for osteoporosis, not only due to corticosteroid therapy but also because of loss of ambulation and muscle strength [5]. All the time research, attention has been directed to developing new therapeutic options to treat DMD, such as exon skipping, gene therapy, and stem cell therapy [6]. Currently, experimental therapy with filgrastim – a granulocyte colony stimulating factor (G-CSF) analogue – is considered to be one of the most promising methods. Due to the fact that it is also a stimulator of stem cells derived from bone marrow, which induces the proliferation of satellite cells and myoblasts as well as muscle regeneration, a significant increase in muscle strength in the limbs has been confirmed in clinical studies [7].
Herein we present a case of boy with a double diagnosis of SRS and DMD. To our knowledge, no similar case has been reported in the literature up to now.
Case report
Our patient is currently a 10-year-old boy born from non-consanguineous, healthy parents.
He was born by caesarean section in the 40th week of gestation, with a birth weight 2100 g (SDS = –4.86 for gestational age), birth length 48 cm (SDS = –1.71 for gestational age), in good condition (8 points in the Apgar scale). He presented characteristic dysmorphic features, such as prominent forehead, relative macrocephaly (head circumference at birth 32 cm, SDS = –1.9 for gestational age), limb asymmetry, 5th finger clinodactyly of both hands, and 2/3 toe syndactyly of both legs feet, suggestive of SRS. Due to dysmorphic features, he was referred to genetic examination in the 17th month, which revealed hypomethylation of the 11p15 region of the paternal chromosome (gene H19). Hence, the diagnosis of SRS was confirmed. The motor development of this boy was delayed; he walked independently in 16th month of life. In the 14th month of life, hepatic dysfunction was suspected due to increased concentration of alanine aminotransferase (AlAT 469 U/l, normal range 0–45 U/l) and aspartate aminotransferase (AspAT 439 U/l, normal range 0–60 U/l). As a result of markedly increased creatine kinase level (21 470 U/l, normal range 0–154 U/l), the diagnosis of Duchenne muscular dystrophy was considered. A genetic test was performed in the 21st month of life. which confirmed the diagnosis because the mutation of the maternal DMD gene located on the X chromosome, encoding dystrophin (exon deletion 49–50), was detected. It was assumed that DMD was diagnosed in asymptomatic phase; however, shortly afterwards health problems typical for DMD appeared. The child was significantly often falling, tiptoed, and had Achilles tendon contractures. Moreover, Gover’s syndrome was positive, and his calves became enlarged.
According to the recommendations, to stop the progression of DMD, the boy has been treated with corticosteroids since he was 4 years old, initially with prednisone (5 to 6 mg/day) and with deflazacort (6 mg/day) for 2 years (i.e. from 8 years of age). During the boy’s treatment with prednisone a significantly slower height velocity was observed, reaching the maximum of 3.3 cm/year.
The patient was qualified for G-CSF treatment within the clinical trial when he was 6 years old. In first year of treatment G-CSF was administrated for 5 consecutive days during first, second, third, sixth, and twelfth month in a dose of 5 µg/kg/day. The treatment resulted in elongation of the distance covered in the 6-minute walk test (6MWT) from 334/385 metres to 330/420 metres. In the subsequent years, the patient received G-CSF 2 or 3 times a year, each time for 5 consecutive days. Up to now, he has received 15 courses of G-CSF treatment.
Moreover, when he was 6 years of age, he was hospitalized due to sepsis, and he required treatment in the Intensive Care Unit. There he experienced severe hypoglycaemia, with neurological symptoms and measured glucose level of 14 mg/dl (0.78 mmol/l). Furthermore, since infancy the patient has been under urological care due to hypospadias. During the last year he underwent cardiological diagnostics because of episodes of sinus tachycardia and heart murmur (2/6 degree in Levine scale). Cardiomyopathy was excluded; however, the cause of those symptoms is still unexplained. Due to tachycardia, the boy is now being treated with propranolol (15 mg per day in 3 equal doses of 5 mg).
From early childhood a significant deficiency of height and body weight were observed (Fig. 1). Therefore, he was diagnosed in the Department of Paediatric Endocrinology and Diabetology. Growth hormone deficiency was excluded (maximum secretion of growth hormone in the sleep test was 31.6 ng/dl); however, the boy fulfilled the criteria for treatment with rhGH in the therapeutic program for short stature children born small for gestational age. The necessary tests were performed when he was 8 years old. At admission, the physical examination revealed dysmorphic features typical for SRS including slightly limb asymmetry, hypotonia, malocclusion, and hypospadias. Pubertal status was assessed as 1 degree in the Tanner scale. Auxological examination revealed the following: height (Ht) 115.9 cm (HtSDS = –2.98), weight (Wt) 19.9 kg, and body mass index (BMI) 14.8 kg/m2 (BMI SDS = –0.6). Because the mother’s height is 170 cm and the father’s height is 182 cm, the target height was calculated to be 182.5 cm (target height SDS = 0.61). The height velocity at the time of diagnosis was 4.3 cm/year. Glucose and insulin levels in oral glucose tolerance test (OGTT) were proper (Table I). IGF-1 and IGFBP-3 concentrations were checked: respectively, 231 ng/ml (normal range: 64–345) and 7.29 ug/ml (normal range: 1.6–6.5). MRI of the hypothalamic-pituitary region showed a transparent septum cyst, but it was not clinically relevant. The treatment was started when the patient was 8 years and 11 months old.
Figure 1
Change of height during life and rhGH treatment. The arrows indicate the position in the growth percentiles by bone age
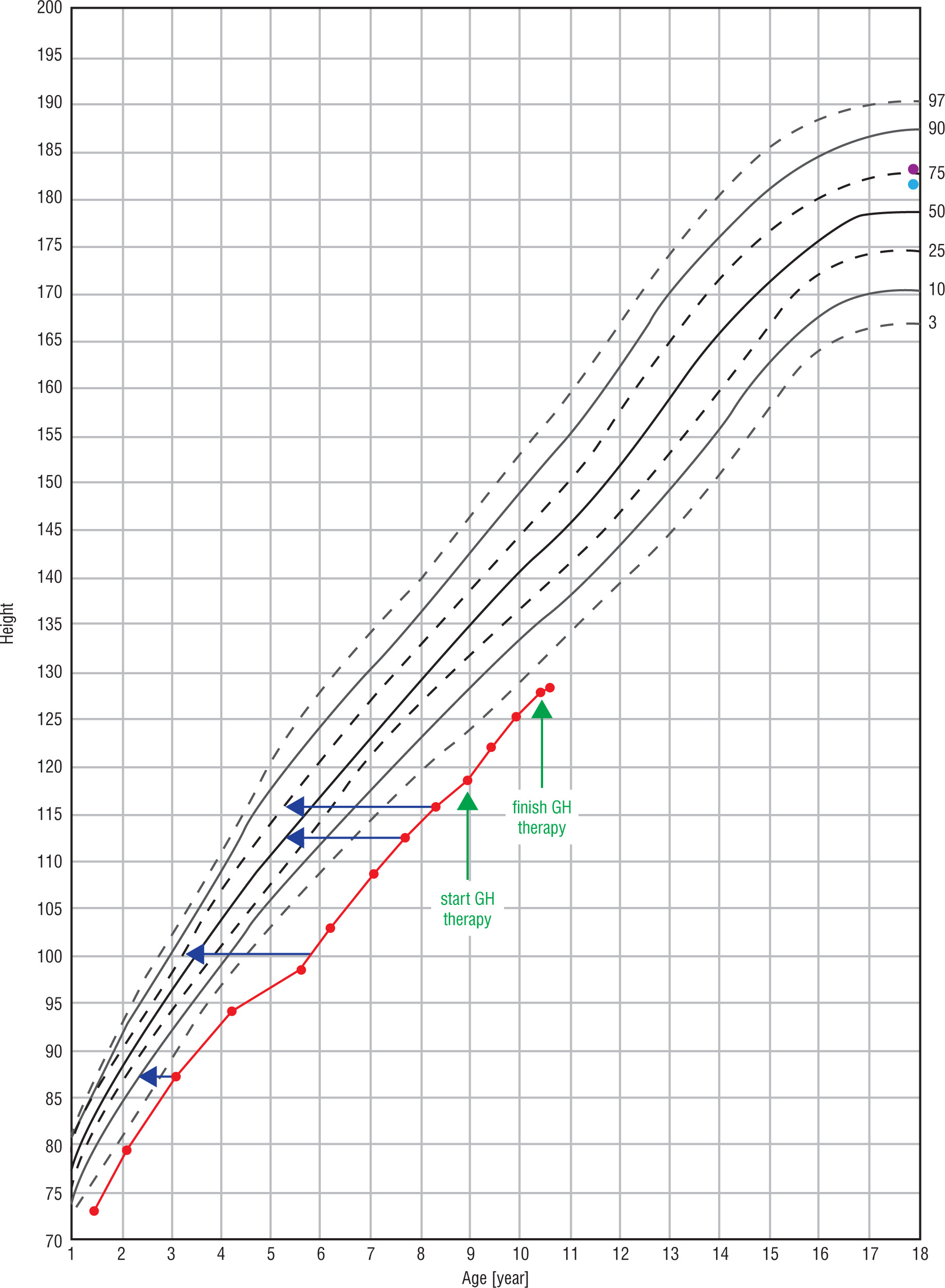
Table I
The result of OGTT performed before and after 1 year of rhGH treatment
| Time (minutes) | 0 | 60 | 120 | |
|---|---|---|---|---|
| Before rhGH treatment | Glucose [mg/dl] | 77 | 91 | 83 |
| Insulin [µIU/ml] | 10.1 | 35.8 | 37 | |
| After 1 year | Glucose [mg/dl] | 86 | 146 | 156 |
| of rhGH treatment | 45.5 | 178 | 375.1 |
He was treated with rhGH for 18 months – in first year of the therapy with a dose of 0.035 mg/kg/per day. During follow-up an improvement of auxological parameters and height velocity was observed. At the start of the treatment Ht SDS was –2.92, after 18th months of the therapy the HtSDS was –2.39 (∆ Ht SDS +0.53). The height velocity during the therapy increased from 4.3 cm/year before the treatment to 6.9 cm/year at 12 months of the therapy. The level of IGF-1 increased, exceeding the upper limit of the norm. It is worth noticing that in the course of treatment the boy’s BMI increased from 16.6 kg/m2 (BMI SDS = –0.18) to 21.9 kg/m2 (BMI SDS = 1.43). Moreover, during the follow-up worsening of glucose metabolism and increasing insulin resistance was observed: at the age of 8 years (while completing results necessary for start the treatment) both glucose and insulin levels in OGTT were normal, HOMA-IR 1.9; at the day of initiation of the treatment HOMA-IR increased to 2.8 and HbA1c was elevated (6.6%). After 1 year of the GH treatment HbA1c was 6%. The result of OGTT empowers the diagnosis of impaired glucose tolerance (IGT) and insulin resistance (HOMA IR 9.66), as presented in Table I.
Furthermore, it should be noticed that during the rhGH treatment the distance covered in 6MWT decreased from 303/348 m to 315 m, and an abnormal, weakened gait was noticed. After 1 year of the treatment, due to rapid weight gain and impaired glucose tolerance, behavioural treatment with lifestyle modification was recommended and the rhGH dose was reduced to 0.03 mg/kg. During the next 6 months the boy grew 2.8 cm. After 18 months of treatment rhGH therapy was discontinued.
The patient’s characteristics including auxological and hormonal evaluation are presented in Table II and Figs. 1 and 2.
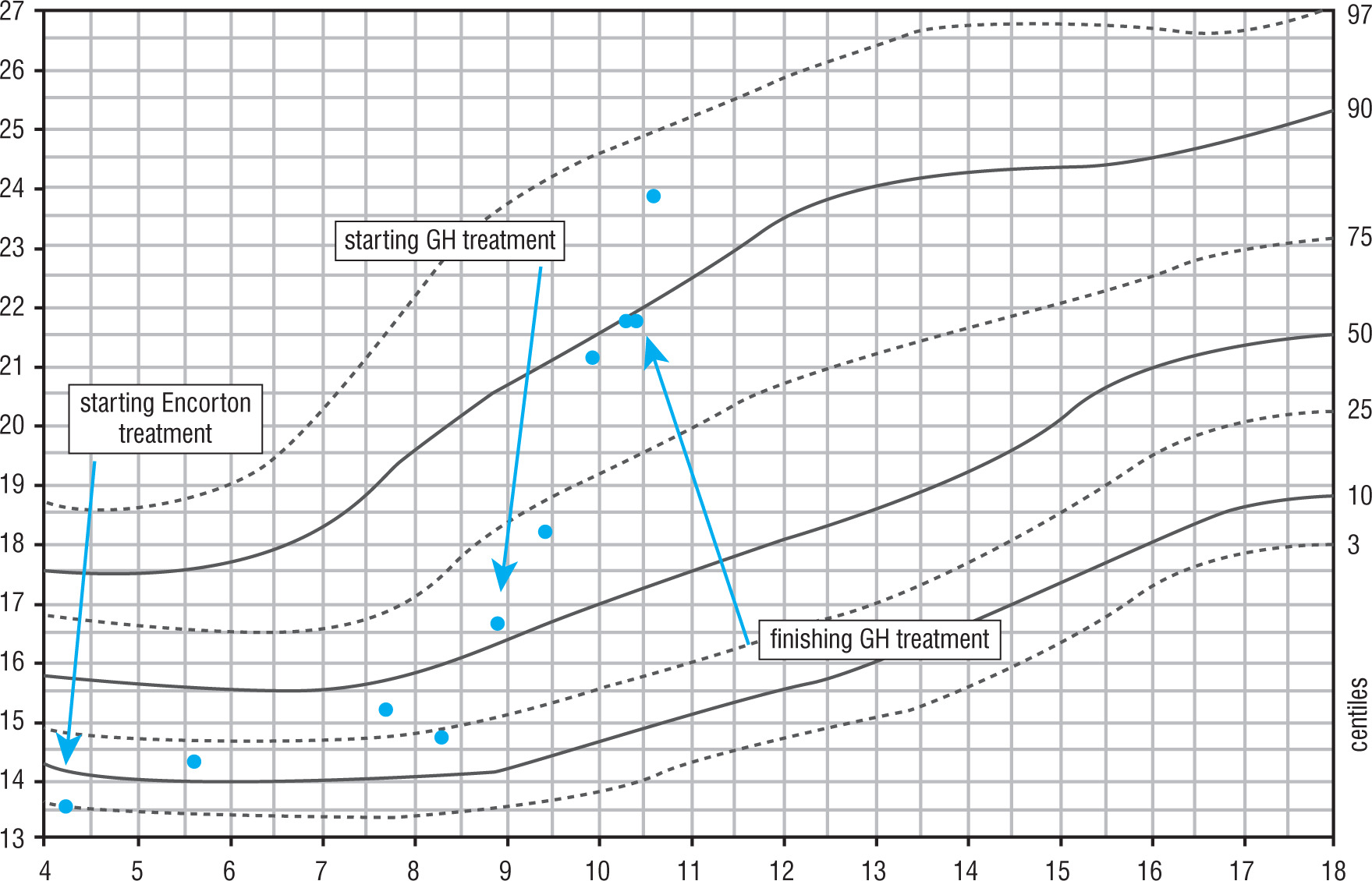
Table II
Patient’s characteristics before the treatment and during the first year of rhGH therapy
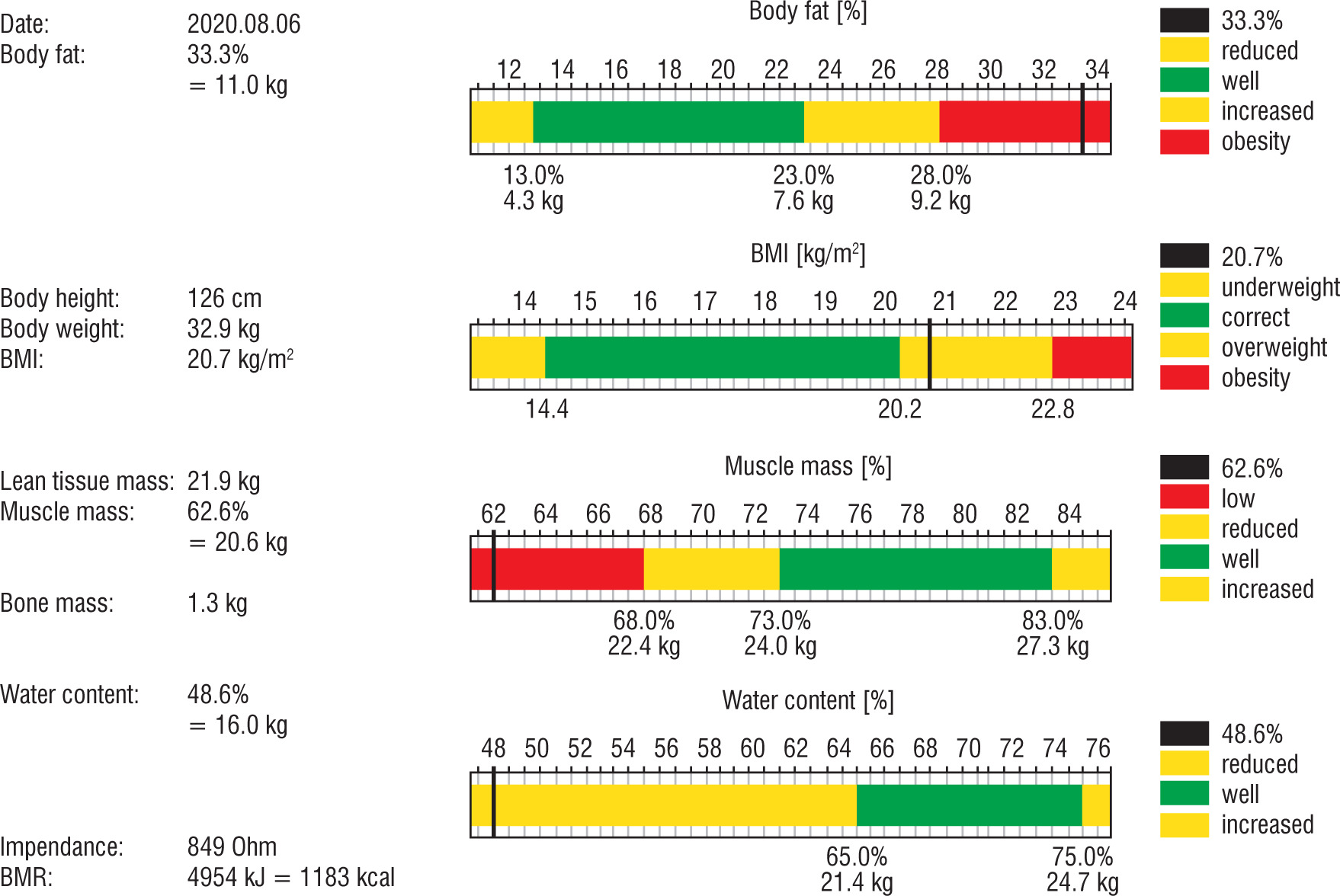
After 1 year of rhGH therapy, densitometry was performed and the body mass composition was examined using the bioimpedance method (Fig. 3). The result of the bone density test of the lumbar spine (L1–L4) was below the age norm (Z-score [–] 2.2).
Discussion
The coexistence of 2 genetic diseases can mutually modify their course. Silver Russel Syndrome is characterized by intrauterine (SGA) and postnatal growth retardation; however, patients may benefit from rhGH treatment. The obvious goal of this therapy is to improve final height. Short stature is also a common manifestation of DMD. In addition, these patients are treated with glucocorticoids in order to slow down the progression of disease; however, such treatment not only exaggerates growth retardation, but is associated with several other adverse effects like weight gain, insulin resistance, and osteoporosis. A significant reduction of growth velocity was also observed in our patient, especially during prednisolone treatment, which resulted in severe growth failure. In Poland, patients with short stature due to SGA, including children with SRS, may be treated with rhGH as part of the reimbursed therapeutic program from 2015. Our patient fulfilled the criteria and qualified for this treatment. However, the question remained whether rhGH therapy in a child with both SRS and DMD would alleviate or worsen the course of muscular dystrophy.
Rutter et al. showed that during 1 year of rhGH treatment in boys with DMD and glucocorticoid-induced growth failure improved the height velocity without detrimental effects observed on neuromuscular function. Growth hormone, in addition to the obvious increase in height, also has a positive effect on muscle mass and strength, through its anabolic effect. Other potential benefits of rhGH treatment are improvement of appetite, and increase in lean body (fat free) mass and bone mass, which can result in improved mobility [8]. It may be assumed that the anabolic effect of rhGH can slow down the progression of DMD. On the other hand, no studies have been large enough to establish whether rhGH therapy has a positive effect on muscle function in DMD.
Duchenne muscular dystrophy is caused by absent or insufficient function of a dystrophin – the cytoskeletal protein that enables the strength and stability of myofibers. It is considered that short statured boys have a higher strength-to-weight ratio, which results in slower progression of DMD and better prognosis; therefore, theoretically, in DMD tall stature might be detrimental to muscle function [9]. This hypothesis is explained by the fact that progression of DMD depends, among others, on the diameter of the muscles, so as long as children are young, short and thin, they may be asymptomatic. Furthermore, the Karpaty concept state that muscles with a larger surface area cause more extensive sarcolemma stress, leading to more severe fibre damage [10].
Molecular testing enables stratification of patients with SRS into subgroups, also with some differences in the clinical picture. One of them is the level of IGF-1. In patients with hypomethylation of the 11p15 region, as is the case in our patient, IGF-1 concentrations often rise significantly above the reference range when treated with standard doses of rhGH, in contrast to children with upd (7) mat or submicroscopic chromosome rearrangements. This also occurred in the presented boy. In addition, elevated levels of IGFBP-3 have been frequently observed in such patients, including ours. Other important clinical difference observed in patients with hypomethylation of the 11p15 region is lower muscle mass, while for patients with upd (7) mat muscle mass is almost normal. Unfortunately, in our patient neither the assessment of body mass composition nor densitometry were performed before the start of rhGH therapy. In body mass composition evaluated after 1 year of the treatment, reduced muscle mass and high content of fat tissue was found, as expected. Rutter et al. showed that during 1 year of rhGH treatment in 39 boys with DMD and glucocorticoid-induced growth failure, lean body mass increased and BMI decreased due to the increase in height [8]. Our observation contradicts this report because during the one-year follow-up, a significant increase in BMI was noticed. However, the rise of body mass was seen before the rhGH therapy was initiated, although it escalated in the second half-year of the treatment. However, we do not associate the weight gain with the use of growth hormone but with a significant change of lifestyle due to coronavirus pandemic. From the pandemic announcement in March 2020 and the recommendation of social distancing, the boy led a highly sedentary lifestyle, not only without school and rehabilitation but also with substantial reduction of any other physical activity, while he consumed more food. It should be stressed that obesity in boys with DMD is very harmful, significantly limiting their functionality and motility, mainly due to the use of residual muscle strength to carry excess fat. Therefore, the recent weakness of gait and the shortening of 6MT may be explained by the patient’s overweight; nevertheless, it may be related to the natural course of muscular dystrophy. However, the decision of discontinuation of rhGH therapy was made due to progressive muscle weakness according to the suggestion of a paediatric neurologist.
Young children with SRS (under 5 years of age) are at risk of fasting hypoglycaemia. The incidence of low glucose level in these children is approximately 27% [1]. In the presented patient the hypoglycaemia was not stated before 6 years of age. This occurred during sepsis with fever, vomiting and diarrhoea, suggesting other possible pathomechanism of lowering of blood glucose.
Glucocorticoid therapy predisposes to disturbances of carbohydrate metabolism due to increase hepatic gluconeogenesis and cellular resistance to insulin. Steroids cause predominantly postprandial hyperglycaemia [11]. Also, growth hormone increases hepatic gluconeogenesis and glycogenolysis, reduces peripheral glucose utilization, and enhances insulin resistance, which results in hyperinsulinaemia and might accelerate the onset of diabetes in patients with pre-existing risk factors for diabetes [12]. Insulin resistance is common in boys with DMD and unfortunately progresses with GH therapy. In a group presented by Rutter et al. the fasting insulin level was significantly higher after 1 year of GH therapy, and 2 boys (5%) developed significant insulin resistance, including 1 who developed impaired glucose tolerance [8]. In our patient, before the initiation of the rhGH therapy, but on the deflazacort treatment, only a slightly increased HbA1c was observed, while after a year of rhGH administration with the high dose used in SGA together with deflazacort, the boy developed IGT and insulin resistance. The weight gain also contributed to the deepening of disturbances of the glucose metabolism. One of the side effects of somatotropin therapy is the acceleration of the progression of scoliosis, included in the spectrum of SRS (9–36% patients) and DMD symptoms. The rate of progression of scoliosis varies, depending, among others, on the period of growth, being greater in the periods of accelerated growth velocity, including rhGH therapy. Fortunately, our patient did not develop scoliosis during the follow-up.
Growth hormone, in addition to the obvious increase in growth, also has a positive effect on bone metabolism. Key recommendations for DMD comprise regular assessment of bone mineral density (BMD) by dual-energy X-ray absorptiometry, starting at initiation of GC therapy. Unfortunately, the available data show that only a minority of patients (24% in the USA) are examined regularly [13]. To our knowledge, the BMD was assessed for the first time in our patient, despite 6 years of glucocorticoid treatment. As expected, the result of the bone density test of the lumbar spine (L1–L4) was below the age norm (Z-score [–] 2.2).
Summary
All available therapeutic options that may benefit patients should be used by physicians. Close monitoring of treatment and side effects, as well as the immediate decision to withdraw from therapy in the event of alarming symptoms, are essential in the case of non-standard therapy. Conducting appropriate behavioural therapy and physical rehabilitation is also necessary to achieve the assumed goal of pharmacological treatment.









