
Introduction
IgG4-related disease (IgG4-RD) is an increasingly diagnosed immune-mediated condition. The pathophysiology of the IgG4-RD is still not fully understood. The disease is associated with chronic inflammation of various organs, which results from their infiltration by activated lymphocytes and plasma cells, with an elevated percentage of IgG4-positive cells [1]. Chronic inflammation is usually accompanied by fibrosis of the affected tissues and organs, obliterative phlebitis, and eosinophilic infiltration. Histopathological examination demonstrates no signs of necrosis or granuloma formation.
The number of patients with IgG4-RD is hard to estimate because the diagnostic criteria for this disease have been proposed quite recently. According to Japanese researchers, the average number of newly diagnosed cases corresponds to 0.28–1.08 per 100,000 population [2]. The disease may also develop in childhood, but published case reports of IgG4-RD in children are sparse. The clinical presentation of the disease is highly heterogeneous and depends on the affected organ [3, 4]. The diagnostic criteria of IgG4-RD proposed in 2012 are based on the location of affected organs and their type of dysfunction, histopathological presentation, and serum concentration of IgG4 [5]. Reliable data from clinical trials that would unequivocally confirm the accuracy of the diagnostic standards are still unavailable; nevertheless, the diagnosis of IgG4-RD established based on those criteria is likely to be correct.
Clinical features suggestive of IgG4-RD include unilateral swelling of lacrimal, submandibular, or parotid gland, sclerosing cholangitis, pancreatitis, thyroiditis, pseudotumor located in the orbit, paranasal sinuses or abdominal cavity, mediastinal fibrosis, sclerosing mesenteritis, inflammation of the aorta, meningeal hypertrophy, interstitial nephritis, and pneumonitis [6].
The conclusions of an international symposium that took place in Boston in October 2011 suggest that diagnosis should be based on the combined features (histopathological and increased numbers of IgG4+ plasma cell). A consensus statement on the pathology contains the scheme based on characteristic histological features such as: dense lymphoplasmacytic infiltrate; fibrosis – storiform in character – and obliterative phlebitis. Depending on the number of pathology features and numbers of IgG4+ plasma cells, the diagnosis of IgG4-RD is histologically highly suggestive or probable [7, 8].
Also, specific laboratory abnormalities (hypergammaglobulinaemia, hypocomplementaemia, eosinophilia) have been defined. It should be emphasised, however, that the serum IgG4 concentration in patients is not necessarily elevated and occurs only in 40% of them (Table 1) [5].
Table 1
Diagnostic criteria [5]
Treatment of IgG4-RD is not always necessary. In asymptomatic cases where no critical organs are involved, the treatment can be postponed and replaced by watchful waiting. All patients with an active form of the disease should be treated. The treatment of choice is glucocorticoid therapy – prednisone [9–11].
Case report
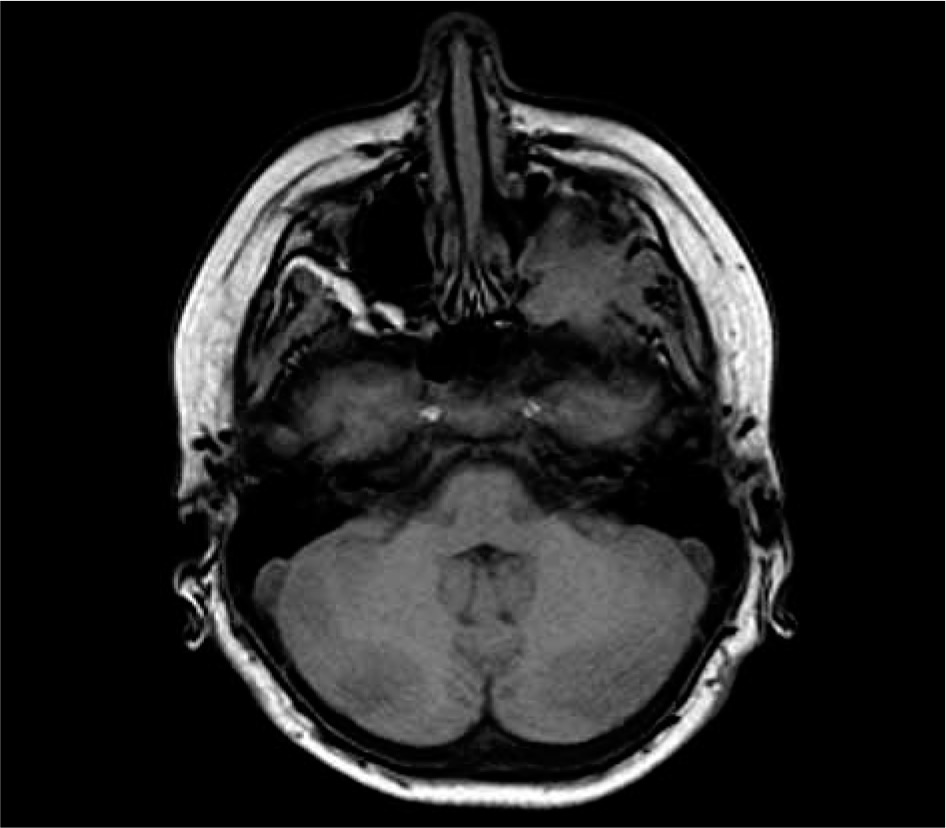
We present the medical history of a boy, currently 13 years old, who has been hospitalised multiple times in different medical centres due to repeated episodes of fever and respiratory infections since he was three years old. The first hospitalisation was associated with a severe bacterial infection: meningitis, pneumonia, and pericarditis. Several subsequent hospitalisations were due to fever of unknown origin. When the patient was seven years old, the symptoms exacerbated in the form of persistent fever and weakness, which resulted in another hospitalisation. The boy presented with anaemia that required transfusion of erythrocyte concentrate – physical examination revealed liver and spleen enlargement; laboratory tests revealed raised inflammatory markers, hypergammaglobulinaemia and elevated concentrations of IgG 2978 mg/dl; the IgG4 concentration was not assessed. The fact that the patient did not respond adequately to empirical wide-spectrum antibiotic therapy raised suspicion of an autoimmune disorder. He received glucocorticoid therapy ordered in another hospital, which resulted in the resolution of fever and normalisation of inflammatory markers. The glucocorticoid treatment was continued for six weeks. Four months after the treatment he started complaining about headaches, and left exophthalmos was observed on clinical examination. MRI revealed a mass around the posterior wall of the left orbit, involving the lateral rectus muscle, sphenoid wing, and masseters (Fig. 1). The image obtained from the test did not indicate a malignant tumour or demyelinating background. Available medical records do not confirm that other imaging examinations were performed. Unfortunately, histopathological examination of endoscopically collected biopsy specimens was non-diagnostic and suggests pseudotumour. The patient received glucocorticoid therapy again, which had been continued for four months. Control head MRI scan after the treatment showed progression of the tumour, which also involved the left pterygopalatine fossa. Earlier available medical documentation does not provide more detail about the diagnosis and treatment at that time.
At this point of time, nine years since initial manifestation of the disease, the patient was admitted to the Paediatric, Haematology, Oncology, and Rheumatology Unit of the District Paediatric Hospital in Bydgoszcz.
The boy underwent tumour surgery: first it included an open surgical biopsy of the tumour of pterygopalatine fossa, while histopathological examination of the specimens showed the characteristics of chronic inflammation. The result of histopathological examination did not allow determination of the aetiology of the inflammation. Five months later, enlarged lymph nodes were found on the neck and in the left supraclavicular fossa. The patient was qualified for another surgery, and some enlarged lymph nodes were collected for microscopic examination during the procedure. Histopathological analysis revealed reactive follicular hyperplasia; sinus histiocytosis of lymph nodes, and excess fibrous connective tissue, with signs of fibrosis and chronic diffuse infiltration of connective and adipose tissue; the pathologist suggested abnormal fibrosis IgG4 (+). Laboratory tests showed significantly elevated serum concentration of IgG 2528 mg/dl (N: 750–1650 mg/dl), with an increase in IgG4 fraction – 350 mg/dl (N: 4–230 mg/dl).
Based on these findings and the results of the histopathological examination of lymph node biopsy specimens, the patient was eventually diagnosed with possible IgG4-RD.
He received prednisone 0.6 mg/kg, which has been continued for three months. Then prednisone dose was slowly tapered to 5 mg. The boy did not tolerate the treatment well; although the fever resolved, he complained about malaise and lower extremity pain.
After one week of treatment with 5 mg prednisone, the patient again presented with fever, bone and joint pain, and elevated inflammatory markers. Hence, the prednisone dose has been maintained at 10 mg/day which resulted in adequate control of the symptoms and stabilisation of serum IgG4 within the normal range. Long-term course, dependence on corticosteroid therapy, lack of histopathological features of malignancy (pathologist suggested IgG4-RD), as well as complete regression of infiltrates in the pterygoid-palatal fossa and lymph nodes confirms our diagnosis of probable IgG4-RD according to Boston criteria and possible diagnosis based on criteria from 2012 [5, 7].
Discussion
Taking into account the heterogeneity of clinical forms and non-specific manifestations of IgG4-RD, physicians of various specialties should consider it as a differential diagnosis in patients suspected of other conditions. A recent systematic review of papers published in 2010 – 2015 identified only 34 out of 740 studies that described IgG4-RD in paediatric patients, among them 25 case reports of children between 22 months and 17 years of age, who were diagnosed with this condition [13]. IgG4-RD should be definitively considered as a differential diagnosis in children with inflammatory pseudotumor of unexplained aetiology located in critical organs: eyeball, lymph nodes, salivary glands, or pancreas [12–16].
The gold diagnostic standard is histopathological examination, while clinical findings, and results of serological tests and imaging studies can be only helpful in directing further diagnostics. The diagnosis of IgG4-related disease requires collaboration between the pathologist and the treating physician. This dialogue is critical in excluding the variety of other diseases that may show elevated serum and tissue levels of IgG4. The isolated presence of IgG4+ plasma cells or an elevated IgG4-to-IgG ratio constitutes relatively non-specific findings. Elevated serum IgG4 concentration is not a specific feature of IgG4-RD, because it is also found in chronic inflammation, infections, cancer, autoimmune disease, vasculitis, and in drug hypersensitivity. One should remember that the proposed terminology scheme for diagnosing IgG4-related diseases is based primarily on the histopathological features. Tissue IgG4 numbers and IgG4:IgG ratios are secondary. The guidelines proposed in this statement do not replace careful clinical-pathological correlation and sound clinical assessment [7, 8].
Untreated IgG4-RD may cause irreversible damage to affected organs. Hence, early, intensive glucocorticoid therapy is recommended. The first-line agent should be prednisone with an initial dose of 0.5–1 mg/kg/day in adults and 1.0–2.0 mg/kg/day in children; however, the dosing can be modified depending on the severity of the symptoms and the aggressiveness of the disease. The treatment with the initial dose should be continued for at least 2–4 weeks, and then the dose should be reduced to the lowest that still provides control of the disease; it is recommended that the therapy last approximately three years [9, 10, 14].
It needs to be emphasised that while prednisone monotherapy is effective in 83% of patients, its persistent clinical effect was observed in only 43%. In 25% of the patients, the symptoms recurred once the prednisone dose was reduced. Another alternative is combination therapy. A combination of Mycophenolate mofetil with azathioprine and methotrexate was shown to be effective in about 50% of the cases [8, 17, 18].
A growing body of evidence suggests that IgG4-RD can be successfully controlled with rituximab, especially in patients in whom glucocorticoid therapy was insufficient [11, 15, 16, 18]. Retrospective analysis of the presented case showed a history of multiple clinical manifestations, including meningitis of unknown aetiology, interstitial pneumonitis, orbital tumour, inflammatory pseudotumours, twice-normal serum concentration of IgG4, and characteristic histopathological presentation. Based on these findings, we eventually determined the diagnosis of IgG-RD. Prednisone therapy produced the desired clinical effect.






