
Introduction
The PI3K/AKT/mTOR pathway is frequently altered in cancer, promoting growth, proliferation and survival, so targeting its three main nodes (PI3K, AKT and mTOR) represents a key therapeutic opportunity [1, 2]. PI3Ks are organized in three classes according to their structural similarities and their activation mechanism; the most studied are class I and are activated by growth factor receptors [3]. There are four class I PI3K isoforms (PI3Kα, β, γ, δ), three class II PI3K isoforms (PI3KC2α, C2β, C2γ) and a single class III PI3K. The four class I isoforms synthesize the phospholipid PIP3. PIP3 is a “second messenger” used by many different cell surface receptors to control the movement, growth, survival and differentiation of cells. Class III PI3K synthesizes the phospholipid PI3P, which regulates endosome-lysosome trafficking and the induction of autophagy, the pathways involved in the destruction of pathogens, the treatment of antigens and the survival of immune cells. Much less is known about the function of class II PI3Ks, but new evidence indicates that they can synthesize PI3P and PI34P2 and are involved in the regulation of endocytosis [4].
PI3K are heterodimers composed of regulatory (p85) and catalytic (p110) subunits and exist in 4 isoforms (α, β, δ and γ) [5]. In cancer, p110a is the major isoform required for transformation with oncogenes [6]. This tumor dependence at p110a may be partially explained by its specific activity, which is considerably greater than that of p110b [7].
PI3K catalyzes the phosphorylation of the 3’OH group on phosphoinositides at the cytoplasmic membrane, inducing Ser/Thr AKT kinase translocation. Once recruited into the plasma membrane, AKT is activated by PDK1- and mTOR2-dependent phosphorylation events [8]. Activation of PI3K signaling is common in human cancers [9].
Aberrant activation of the PI3K/AKT/mTOR pathway at different levels of signaling is frequently observed in many human malignancies [10]. The inhibition of PI3K signaling is effective in the treatment of several types of cancer. Intrinsic and acquired resistances limit the therapeutic efficacy of PI3K inhibitors [11].
Various activating mutations in oncogenes as well as the inactivation of tumor suppressor genes are found in various malignant tumors in almost all members of the pathway. Substantial progress in the discovery of PI3K/AKT/mTOR alterations and their roles in tumorigenesis have allowed the development of new targeted molecules with potential to develop an effective anticancer treatment. Two approved cancer drugs, everolimus and temsirolimus, illustrate targeted inhibition of PI3K/AKT/mTOR in the clinic and many more are in preclinical development and are being tested in early clinical trials for many different types of cancer [10].
Blocking the PI3K/AKT/mTOR pathway increases antitumor activity [12]. Preclinical data have shown that the combination of compounds simultaneously targeting different related molecules in the PI3K/AKT/mTOR pathway leads to synergistic activity [13]. Mutations or amplifications in RTK, PTEN or RAS are likely to activate PI3K and other important pathways and to cut negative feedback loops. These other pathways must confer a selective advantage and it will therefore be important to close them, in addition to PI3K, to completely inhibit tumor growth. Therefore, it is suggested that these tumors will respond well to PI3K inhibitors in combination with other targeted therapies. Combination therapy also has the potential to overcome drug resistance or escape dependence on oncogenes [14].
Based on these results, we provide an in silico strategy for the exploration of PI3K/AKT and PI3K/mTOR dual competitive ATP inhibitors.
The marked interest in the development of new PI3K/AKT and PI3K/mTOR inhibitors as potential agents for cancer treatment prompted us to explore the possibility of developing these inhibitors on the basis of QSAR models to predict the bioactivity of AKT and mTOR inhibitors towards PI3K and the interaction of the best ones will be evaluated by docking analysis.
Material and methods
Dataset generation
Active inhibitors against PI3K were extracted from BindingDataBase (https://www.bindingdb.org); their IC50 (molecule concentration leading to 50% inhibition) was transformed into the logarithmic scale, pIC50. 140 chemically diverse compounds with high activity with pIC50 greater than 8 were chosen for the present QSAR study. MTOR and AKT inhibitors with pIC50 greater than 8 were also selected in order to predict their activity against PI3K using the QSAR model developed and to explore their double activity. High activity compounds were chosen to predict future effective dual inhibitors.
QSAR model generation
184 2D descriptors available on the MOE 2008.10 (obtained from Chemical Computing Group (CCP); Montreal, QC, Canada) [15] were calculated for the 140 compounds. Invariant and insignificant descriptors were initially eliminated; then the QSAR contingency descriptor selection and intercorrelation matrices between descriptor pairs were used to extract the 64 most relevant molecular descriptors which were employed for the distance calculation of each database entry.
All 140 selected compounds were distributed randomly in the training set with 100 compounds (70% of the data) and test set consisting of 40 compounds (30% of the data).
Partial least squares (PLS) analysis based on the leave-one-out (LOO) method was used to correlate molecular descriptors with pIC50 values.
QSAR model validation
The internal validation procedure evaluates the relative predictive performance of the QSAR model, on the one hand by the correlation coefficient (R2) used to measure the correlation between the experimental pIC50 and predicted interest values in order to observe the variability between the variables in the set test, and secondly by the root mean square error (RMSE), a parameter used to evaluate the relative error of the QSAR model.
The model was also tested by cross-validation using the LOO (leave-one-out) method and the computation of R2 and RMSE. The Z-scores $Z-SCORE and $XZ-SCORE were used to detect the outliers.
External validation consists of evaluating the activities of the predictions and calculating the numerical parameters using the model.
Activity prediction
The QSAR-PI3K model constructed and validated was used to predict the activity of two groups of AKT and mTOR inhibitors against PI3K, first AKT inhibitors and second mTOR inhibitors. These inhibitors extracted from the Binding Database (https://www.bindingdb.org) have a pIC50 greater than 8 with 578 and 1008 inhibitors for AKT and mTOR respectively.
After calculating the predicted activity, the 40 inhibitors with the best predictions in each group were chosen for docking into PI3K to explore their dual activity.
Molecular docking
The 3D coordinates of the mTOR inhibitors as well as the AKT that showed the best predicted activity in the QSAR-PI3K model have been generated from 2D. 5ITD (PDB ID) is the PI3Kγ crystallized structure retrieved from the PDB database with a resolution of 3Å for docking analysis. MGL tools 1.5.6 with AutoGrid4 and AutoDock vina (Scripps) [16] were used for docking studies. The PI3K structure was hydrogenated using MGL Tools and PyMol was used to visualize the results [17].
In this work we adopted the same docking strategy used by the authors in previous studies [18]. The grid boxes were generated around the active site of the two three-dimensional structures of the PI3K kinase protein using MGL Tools 1.5.6. The grid boxes were set to have between 16 and 20Å of edge with coordinates x = 19.698, y = 62.891, z = 20.837. The coordinates were determined using the potential substrate binding residues as centroids (in the hinge region and the activation loop) [19].
Dactolisib and pictilisib, known PI3K inhibitors, were also docked into PI3K and were used as a control to evaluate docking results. Their 3D structure was extracted from PubChem.
Results
QSAR analysis
The PI3K-QSAR model was built on the basis of 64 molecular descriptors. After the 140 molecules’ QSAR regression analysis, a PI3K-SAR model with R2 of 0.81706 and RMSE of 0.16029 was obtained, which was validated and evaluated by the cross-validation method LOO. The predictive performance of this model was represented by cross-validated RMSE with 0.59322 and R2 with cross-validation of 0.61031.
Figure 1 shows a plot of experimental and predicted inhibitory potency in pIC50 values of the training and test set compounds showing a comparable and similar distribution between the two groups. No outliers were observed in the test set data, and all compounds were well predicted with a residual value less than one log unit.
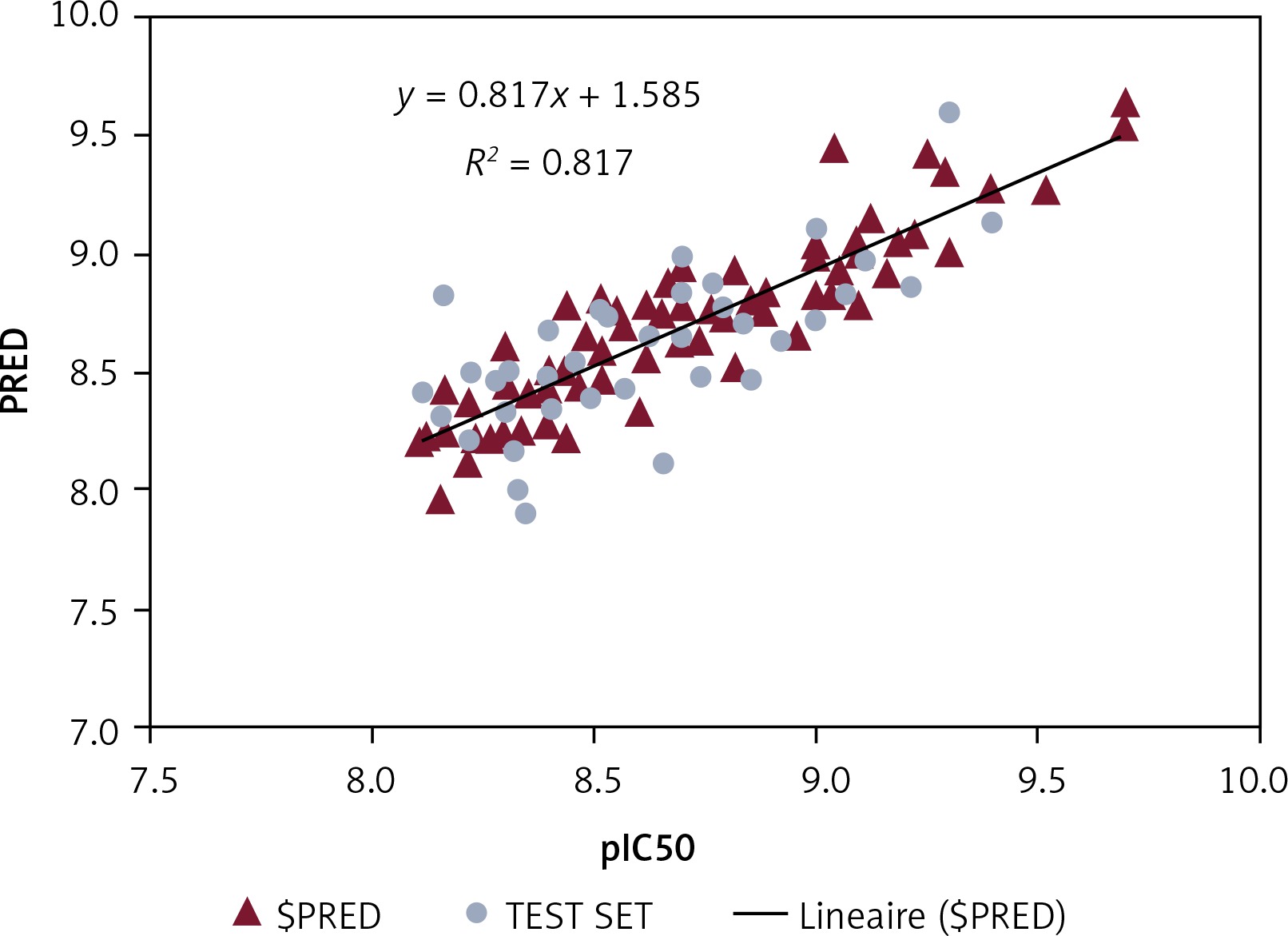
Virtual screening
The PI3K-QSAR model developed and validated was used to predict the activity of AKT and mTOR inhibitors against PI3K. After calculating the predicted activity, the 40 inhibitors presenting the best predictions in each group were chosen to perform their docking into PI3K. The best predicted AKT and mTOR inhibitors present respectively pIC50 activities between 9.26–9.93 and 9.53–9.87.
Molecular docking studies
The docking scores (affinity) of the docked AKT and mTOR inhibitors in the catalytic site of PI3K were between –7.2 and –11.6 kcal/mol for the AKT inhibitor group and between –7.6 and –11.2 kcal/mol for the mTOR inhibitor group; dactolisib and pictilisib (PI3K reference inhibitors) presented respective scores of –7.2 and –8.2 kcal/mol as reported in Tables 1 and 2.
Table 1
Docked interaction analysis of AKT inhibitors screened into PI3K
Table 2
Docked interaction analysis of mTOR inhibitors screened into PI3K
Discussion
The PI3K/mTOR (phosphoinositide 3-kinase/mammalian target of rapamycin) pathway is stimulated by a variety of growth factors and their receptors and regulates cell metabolism, cell growth, cell survival, cell proliferation, cell motility, and angiogenesis. The PI3K/AKT/mTOR pathway is thought to be one of the most frequently mutated pathways in cancer, leading to cancer progression and resistance to existing treatments. New compounds targeting different components of the PI3K/AKT/mTOR pathway continue to be developed.
In order to explore new PI3K/AKT and PI3K/mTOR common inhibitors, a 2D-QSAR model for PI3K was constructed to predict the activity of one group of AKT inhibitors and one of mTOR, then do the docking for those that presented the best prediction (the top 40 in each group) whose results were compared with those of dactolisib and pictilisib. The PI3K inhibitors that were used in the construction of the QSAR model and those of AKT and mTOR whose activity for PI3K was predicted were extracted from Binding Database and selected according to their bioactivity IC50. The model was developed by the PLS method by the MOE software.
The docking score of the AKT and mTOR inhibitors compared to the dactolisib and pictilisib score predicts that the AKT and mTOR inhibitors have an affinity between –7.2 and 11.6 kcal/mol and –7.6 and –11.2 kcal/mol, respectively, while dactolisib and pictilisib have values of only –7.2 and –8.2 kcal/mol, respectively, and therefore all the inhibitors studied had a higher PI3K affinity than dactolisib, and 33 AKT inhibitors and 37 mTOR inhibitors had a higher affinity than that of pictilisib. Thus, the affinity and interaction of pictilisib would be superior to those of dactolisib.
Visualization of these interactions showed that the different compounds adapt to the ATP binding site forming hydrogen bonds, in particular with the residue V851, which is considered as a hinge residue for PI3K; the best AKT inhibitor establishes 7 hydrogen bonds with PI3K, that of mTOR establishes 6 bonds, while dactolisib and pictilisib have respectively only 3 and 4 connections. The important score of the affinity of some inhibitors despite the low number of hydrogen bonds can be explained by the hydrophobic interactions and that of Van Der Waal. Thus, the affinity and interaction of pictilisib and most of the inhibitors studied are more efficient than those of dactolisib, which is also known under the name of NVP-BEZ235 or BEZ2351 as a dual inhibitor of PI3K/mTOR signaling [20, 21]. Dactolisib entered a clinical trial in patients with renal cell carcinoma, but was discontinued due to a high incidence of gastrointestinal side effects [22].
A double comparison of the inhibitors was made with respect to pictilisib which, in consideration of the affinity and the number of PI3K-mediated hydrogen bonds, predicts that 10 AKT inhibitors (Table 3) and 19 mTOR inhibitors (Table 4) both have better results than pictilisib.
Table 3
Comparison of studied AKT inhibitors to pictilisib
Table 4
Comparison of studied mTOR inhibitors to pictilisib
Taking into account the rules of Lipinski (no more than 5 hydrogen bond donors, no more than 10 hydrogen bond acceptors, an octanol-water partition coefficient log P not greater than 5.5) and that of Veber determining the good bioavailability (polar surface area [PSA] ≤ 140 Å2 [absolute polarity measurement], rotatable bonds ≤ 10 [flexibility measurement]), we found that 4 AKT inhibitors (Table 5), of which 50418205 is the most relevant (5 hydrogen bonds and an affinity of –10.1kcal/mol), and 14 mTOR inhibitors (Table 6), of which 270379 is the most relevant (6 hydrogen bonds and an affinity of –11.2 kcal/mol), met all the criteria and could be future drugs (Fig. 2).
Table 5
AKT inhibitors meeting Lipinski and Veber criteria
Table 6
mTOR inhibitors meeting Lipinski and Veber criteria
Fig. 2
Visualization of the different interactions between the compounds and the active site of PI3K via the hydrogen bonds. Dactolisib (A) and pictilisib (B) respectively have 3 and 4 linkages with the catalytic site of pi3K whereas 270379 inhibitor of mTOR (C) and 50418205 inhibitor of AKT (D) respectively have 5 and 6 hydrogen bonds. Notes: red zones: oxygen atom, blue zones, nitrogen atom and green zones, other. The hydrogen bonds are represented by the dashed yellow lines. The numbers represent the size of the hydrogen bonds established between the ligand and the receptor
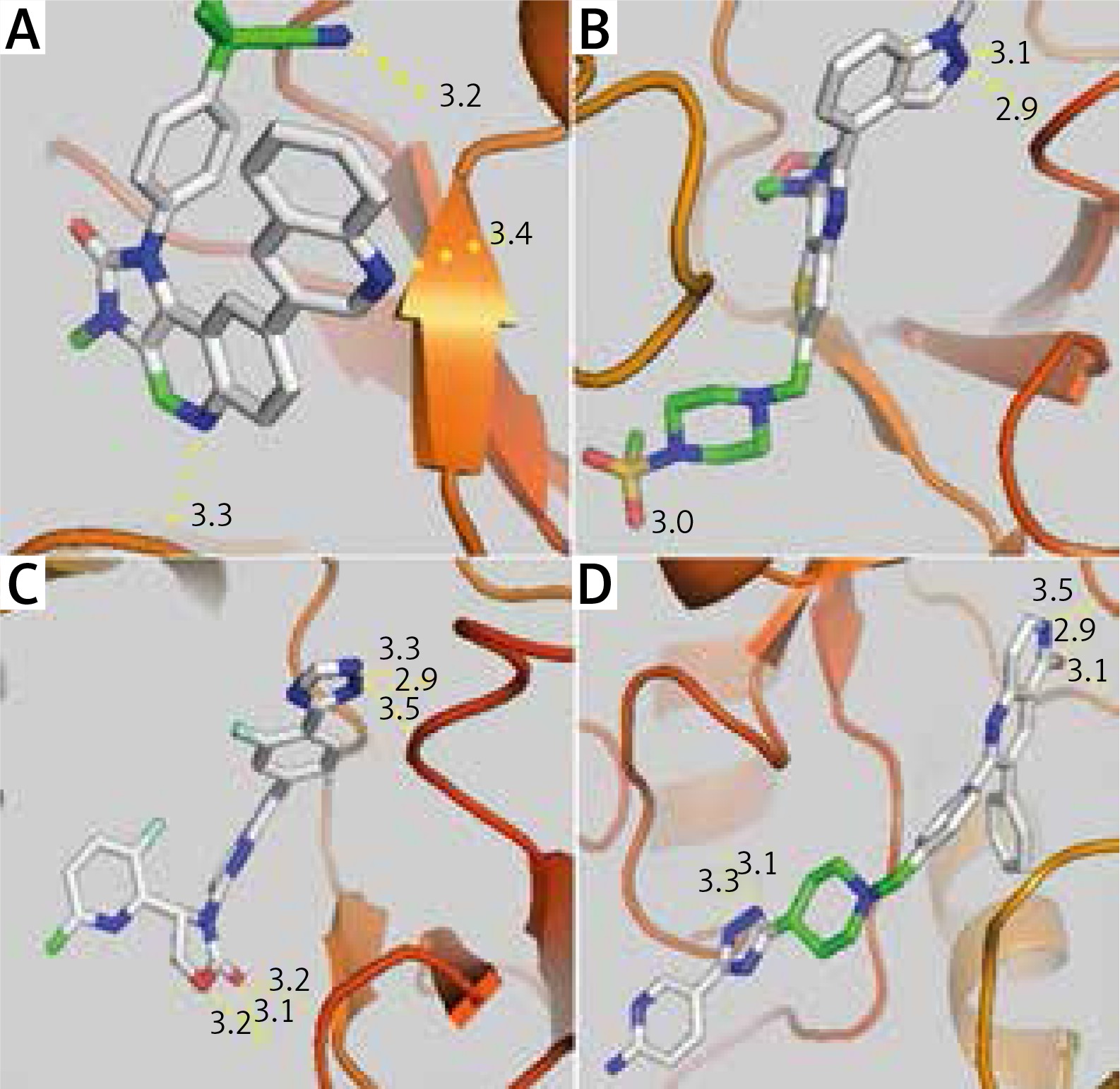
Knowing that some mTOR inhibitors have a partition coefficient log P ≤ 3 and could therefore be better than pictilisib (GDC-0941), a potent and selective oral PI3K [23] class II inhibitor rapidly absorbs after oral administration [24].
Conclusions
The hyperactivation of PI3K/AKT/mTOR in cancer, associated with the crucial role of PI3K signaling in tumorigenesis, has led to significant efforts to generate inhibitors to target this pathway. Through the construction of a QSAR model, we were able to predict potent AKT inhibitors and mTOR activity towards PI3K, select the best ones and anchor them with PI3K while comparing them to PI3K reference inhibitors. Promising results have been obtained; the PI3K/mTOR and PI3K/AKT dual inhibitors have been predicted, pending the testing of these compounds directly on the cancer cell lines in our next study.






