

Introduction
Regulatory T cells (Tregs) play a significant role in preventing development of autoimmune diseases by suppression of activation, proliferation, or differentiation of many subpopulations of immunocompetent cells, including mast cells [1], CD4+ [2] and CD8+ T cells, natural killer cells [3], and B cells [4]. However, it is strongly suggested that the major target of suppressive activity of Tregs are dendritic cells (DCs) [2], as Tregs strongly suppress DCs maturation [5]. Tregs constitute 5-15% of peripheral CD4+ T cells in both mice and humans, which are identified by the expression of interleukin 2 receptor α-chain – CD25 [6, 7] and the nuclear marker Foxp3, which is an essential factor for development and function of Tregs [8, 9].
Natural Tregs originate in the thymus, migrate to lymphoid organs and to the sites of inflammation, where they inhibit T cell proliferation and autoimmune response [10]. This migration is facilitated by the expression of several chemokine receptors [11]. The first evidence that human Tregs express chemokine receptors and respond to stimulation with chemokines was published in 2001 [12]. That study reported the expression of CCR4 and CCR8 on blood-borne human CD4+CD25+ Tregs and their chemotactic response to chemokines CCL22, CCL17, and CCL1 as well as to the viral chemokine vMIP-I [12]. Zou et al. reported that human Tregs accumulate in bone marrow and express CXCR4, which facilitate the migration of CD4+CD25+ Tregs to CXCL12 in bone marrow [13]. It was also shown that blocking of CXCL12/CXCR4 interaction contributes to the reduction of Tregs trafficking to the bone marrow [13]. Moreover, it was shown that 1.25-dihydroxyvitamin D3 regulates T cell differentiation and suppresses IL-17 producing T cells [14]. Other drugs, such as alpha-1 antitrypsin (AAT) [15] and chloroquine [16] increase CD4+FoxP3+ Treg cell population and reduce inflammation in mice, whereas 17β-estradiol can compensate for the lack of Tregs in mice with experimental autoimmune encephalomyelitis (EAE) [17]. Moreover, it was found that mesenchymal stem cells (MSCs) can suppress the proliferation, activation, and differentiation of CD4+ T cells induced to differentiate into Th1 and Th17 cells in mice with EAE [18]. This suppressive effect was associated with generation of CD4+CD25+Foxp3+ regulatory T cell subpopulation [18]. Also, it was also shown that CD4+CD25+CD69- Tregs express CCR7 and efficiently migrate to CCL19, but only a small subpopulation of CD4+CD25+CD69- Tregs express CXCR5 and migrate to CXCL13 [19]. Interestingly, upon activation, Tregs switch the chemokine receptor expression from CCR7 to CXCR5 [19]. More recently, it has been reported that two different functional subpopulations of memory Tregs can be distinguished by the expression of CCR7 [20]. Central memory Tregs (TCM) express CCR7 as opposed to effector memory Tregs (TEM) [20].
Another chemokine receptor expressed by Tregs is CCR6 [21, 22]. CCR6+ Tregs seem to represent “regulatory effector – memory” T cells (TREM) both in mice and humans, because they exhibit a phenotype of activation, expansion, and memory typical for effector memory T cells [21], as opposed to CCR7+ Tregs [20]. CCR6 is characteristic for the relatively new subset of T cells called “Th17 cells” [22, 23]. Th17 cells are found to secret IL-17 and IL-22 [24-26], and play an important role in the induction of EAE [24, 27]. Esplugues et al. suggested that Th17 cells can acquire a regulatory phenotype with in vitro and in vivo immune-suppressive properties (rTh17) in the small intestine of mice with EAE [28].
In this study, we showed that Tregs increasingly migrate to the central nervous system (CNS) during subsequent phases of EAE (preclinical, initial attack, and remission). In peripheral tissues (blood, lymph nodes, and spleen), a significant accumulation of Tregs is present only during remission of EAE. Furthermore, we identified significant up-regulation of CCR6 expression in the CNS, blood, lymph nodes, and spleen in all phases of EAE. The expression of CCR6 on Tregs was the highest during the initial attack of EAE in the CNS and lymph nodes. To analyze further the role of CCR6 on Tregs in EAE, we attempted to modulate disease course by administration of Tregs with blocked CCR6 to EAE mice. Animals receiving these modified Tregs presented significant amelioration of the disease.
Material and methods
Animals
In all experiments, 8-12 weeks old female (SWRxSJL)F1 mice were used. All animals were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and housed at animal facility of the Medical University of Lodz, Poland under standard conditions. All experimental protocols were approved by the Animal Care Committee of the Medical University of Lodz.
Experimental autoimmune encephalomyelitis induction and tissue collection
EAE was induced by active immunization with an encephalitogenic PLP (proteolipid protein) peptide representing residues of 139-151 (PLPp: 139-151, Metabion, Martinsried, Germany) emulsified with complete Freund’s adjuvant (Sigma, Poznan, Poland). Pertussis toxin (Sigma, Poznan, Poland) was injected i.v. at the day of immunization and 48 hours later, as previously described [29]. All mice were weighted and examined daily for clinical signs of EAE. The following clinical scoring scale was used: 0, no disease; 1, decreased tail tone or slightly clumsy gait; 2, tail atony and/or moderately clumsy gait and/or poor righting ability; 3, limb weakness; 4, limb paralysis; 5, moribund state.
For cell analysis during preclinical phase (day 10 postimmunization), the initial attack (the first three days of EAE signs), and remission of the disease, mice were anesthetized with ketamine/xylazine cocktail (100 mg/kg of ketamine and 10 mg/kg of xylazine) (Biowet, Pulawy, Poland) administered intraperitoneally and perfused through the left cardiac ventricle using ice-cold phosphate-buffered saline (PBS, Biomed, Poland) with heparin. Preclinical phase was defined as the time with no EAE signs, initial attack of EAE as the phase when clinical signs appeared (score 1-4), and remission was identified as the time when clinical signs improved. Each investigated group contained 7 to 14 mice.
Isolation of mononuclear cells from the CNS
After being collected from an animal, the CNS (brain and spinal cord) was placed in ice-cold PBS and forced through 70 µm cell strainers (BD Bioscience, Bedford, MA, USA) for a single cell suspension and then centrifuged for 10 min at 350 × γ (4°C). The CNS mononuclear cells were resuspended in 40% percoll (Sigma, Poznan, Poland) and diluted with white Hank’s buffered salt solution (HBSS) (Lonza, Basel, Switzerland). Then, the suspension of cells was carefully overlayered on top of the 70% percoll, diluted with red HBSS. CNS mononuclear cells were isolated by centrifugation for 40 min at 700 × γ (4°C) with slow acceleration without a break. Cells were then collected from the 40%/70% interface, washed and resuspended in PBS [30]. In the next step, the CNS cells were stained with trypan blue solution 0.4% (Sigma, Poznan, Poland) and counted in the Bürker chamber (Merck, Warsaw, Poland) under a light microscope objective 10x (Carl Zeiss, Axio Observer.A1, Poznan, Poland) and prepared for flow cytometry.
Isolation of mononuclear cells from blood, lymph nodes, and spleen
After being collected from animals, the analyzed tissues (draining lymph nodes – inguinal and axillary, and spleen) were placed into ice-cold PBS and forced through 70µm cell strainers for a single cell suspension and centrifuged for 10 min at 350 × γ (4°C). Blood mononuclear cells were separated on Histopaque 1077 (Sigma, Poznan, Poland) and samples were centrifuged for 25 min at 400 × γ (25°C). Then, the cells were collected from the interface and washed with PBS. The remaining red cells from the blood and spleen preparation were lysed by hypotonic shock in NH4Cl, KHCO3, and Na2EDTA × 2H<sub>2</sub>O buffer, pH = 7.2 (red blood cells, RBCs buffer; Sigma, Poznan, Poland) for 5 min at 4°C [31]. Later, the cells were washed by centrifugation for 10 min at 400 × γ (4°C) and resuspended in PBS. Cells isolated from the blood, lymph nodes, and spleen were stained with 0.4% trypan blue solution, and counted in Bürker chamber under a light microscope (objective 10×) and prepared for flow cytometry.
Analysis of CCR6 expression on Tregs by flow cytometry
Mononuclear cells from the CNS, blood, spleen, and lymph nodes (1 × 106) were resuspended in 100 µl of PBS and incubated with FcBlock (BD Bioscience, San Jose, CA, USA) for 15 min at 4°C. After this time, specific rat antibodies for murine CD4-Pacific Blue, CD25-APC-Cy7, and CCR6-Alexa Fluor 647 (BD Bioscience, San Jose, CA, USA) were added at a predetermined optimal concentration for 15 min at 4°C. The cells were washed with 1 ml of PBS and permeabilized according to the manufacturer’s protocol (eBioscience Inc., San Diego, CA, USA; mouse regulatory T cell staining kit, cat. no. 88-8111). FcBlock was added before incubation with Foxp3 antibody. Later, 0.5 µg per test of anti-mouse/rat Foxp3-PE, Foxp3-APC, or Foxp3-PE-Cy7 antibodies (eBioscience Inc., San Diego, CA, USA) were added to each tube. The incubation lasted for 30 min at 4°C in dark. Then, the cells were washed and fixed in 200 µl of 1% formaldehyde/PBS solution and stored at 4°C until further analysis. The analysis was carried out on FACS Canto II instrument (BD Bioscience, San Jose, CA, USA) and data were analyzed using FACSDiva 6.0 software (BD Bioscience, San Jose, USA).
Immunohistochemistry
Animals were transcardially perfused with 4% formalin (Chempur, Piekary Slaskie, Poland). The CNS and spleen were removed, post-fixed for 2 days in 4% formalin solution, and embedded in paraffin. Sections (10 µm thick) were placed onto glass slides (Menzel-Glaser, Braunschweig, Germany). Slides with paraffin-embedded sections were deparaffinized by immersion in xylene (Sigma, Poznan, Poland), followed by rehydration in ethanol (POCH, Gliwice, Poland). Antigen retrieval was done with 10 mM citrate buffer, pH 6.0 (Sigma, Poznan, Poland). Tissue sections were incubated overnight at 4°C, with primary rat anti-Foxp3 antibody (eBioscience Inc., San Diego, CA, USA) at 1:500 dilution. Secondary biotinylated rabbit anti-rat antibodies were purchased from DAKO North America Inc. (Carpinteria CA, USA) and ABC kit from Vector Laboratories (Peterborough, United Kingdom). Staining was performed using diaminobenzidine substrate (Sigma, Poznan, Poland) and counterstaining was done with hematoxylin-eosin (Sigma, Poznan, Poland).
Modulation of EAE using CCR6-deficient Tregs
The cells isolated from drained lymph nodes (inguinal and axillary) of animals with EAE remission (3rd and 4th day) were counted in Bürker chamber under a light microscope objective 10x. Next, Tregs were sorted using CD4+CD25+ regulatory T cell isolation kit, according to the manufacturer’s instruction. Approximately, 4 mln of Tregs were obtained from each mouse. Half of these cells were incubated with anti-CCR6 antibody (R&D Systems, Minneapolis, MN, USA) and the other half was left unchanged. Concentration of anti-CCR6 antibody was 4.5 µg/ml per 1 mln of Tregs resuspended in 100 µl of PBS. Tregs were incubated with antibody for 20 min at 4°C. After this time, 1 ml of PBS was added to the cells and unbound antibody was removed by centrifugation (10 min, 350 × g, 4°C). CCR6 protein expression on Tregs and on Tregs with blocked CCR6 was analyzed by flow cytometry.
Tregs (2 mLn cells) and Tregs with blocked CCR6 (2 mln cells) were separately applied to two different groups of immunized mice during preclinical phase of EAE (7th or 8th day post-immunization). Both groups consisted of 6 mice. Tregs were given slowly, intravenously to the mouse tail vein at a concentration of 1 mln/ml. The third group of animals were the control group and consisted of 5 mice with unmanipulated EAE to show natural course of the disease. All animals were examined daily for 39 days, and EAE scores were provided by two independent observers.
Results
Accumulation of Tregs in the CNS and peripheral tissues during EAE
The number of Tregs (CD4+CD25+Foxp3+) isolated from the CNS increased significantly during all three analyzed phases of EAE (preclinical, initial attack, and remission) in comparison to control unimmunized group (p = 0.03, p = 0.0001, and p = 0.002, respectively; Mann-Whitney test) (Figs. 1 and 2A). During remission of EAE, the number of Tregs accumulating in the CNS was significantly higher than in the preclinical phase and initial attack (p = 0.03 and p = 0.0003, respectively; Mann-Whitney test) (Fig. 2A).
Fig. 1
Flow cytometry analysis of Tregs (CD4+CD25+- Foxp3+ T cells) from the central nervous system (CNS) of control normal animals (A) and from the CNS of experimental autoimmune encephalomyelitis (EAE) animals obtained during three successive phases of the disease: preclinical (B), initial attack (C), and remission (D). Dot plots are shown for the populations gated on CD4+ T cells and display surface staining of CD25 versus Foxp3. Data is representative of individually analyzed mice (n = 14 per group). Numbers represent the percentage of CD25+- Foxp3+ cells. Cells from the CNS of control unimmunized mice were pooled from 4 mice
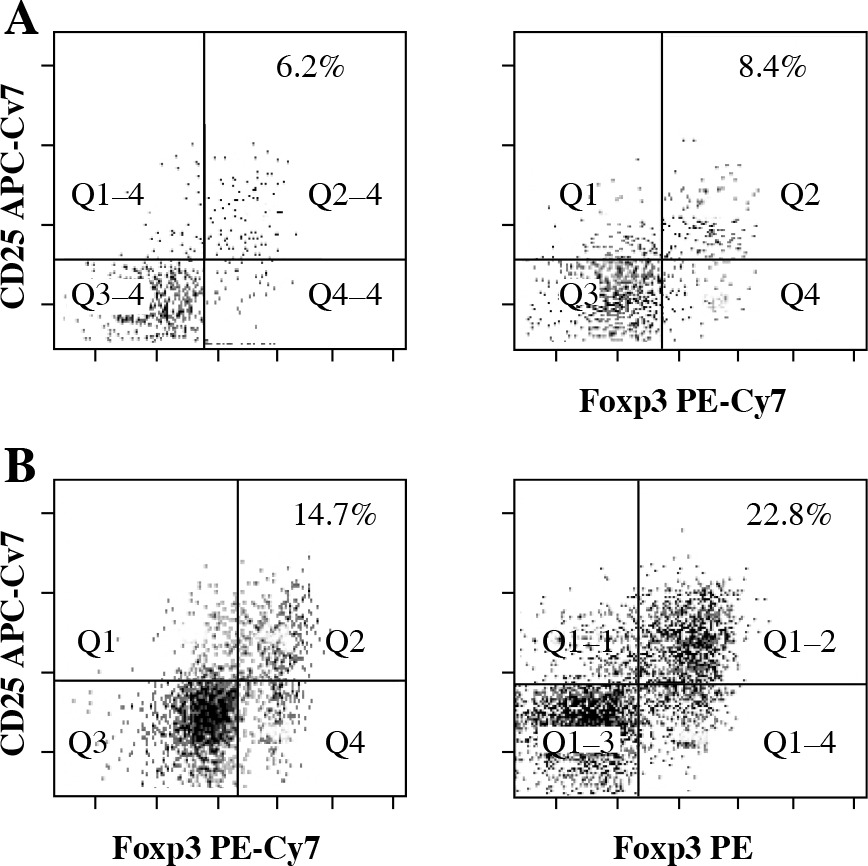
Fig. 2
Accumulation of Tregs during three consecutive phases of experimental autoimmune encephalomyelitis (EAE) (preclinical, initial attack, and remission) in the central nervous system (CNS) (A), blood (B), lymph nodes (C), and spleen (D) analyzed by flow cytometry. Cells from the CNS of control unimmunized mice were pooled from 6 mice. Data is derived from a CD4+ cell gate, n = 14 per group, Mann-Whitney test with Bonferroni correction
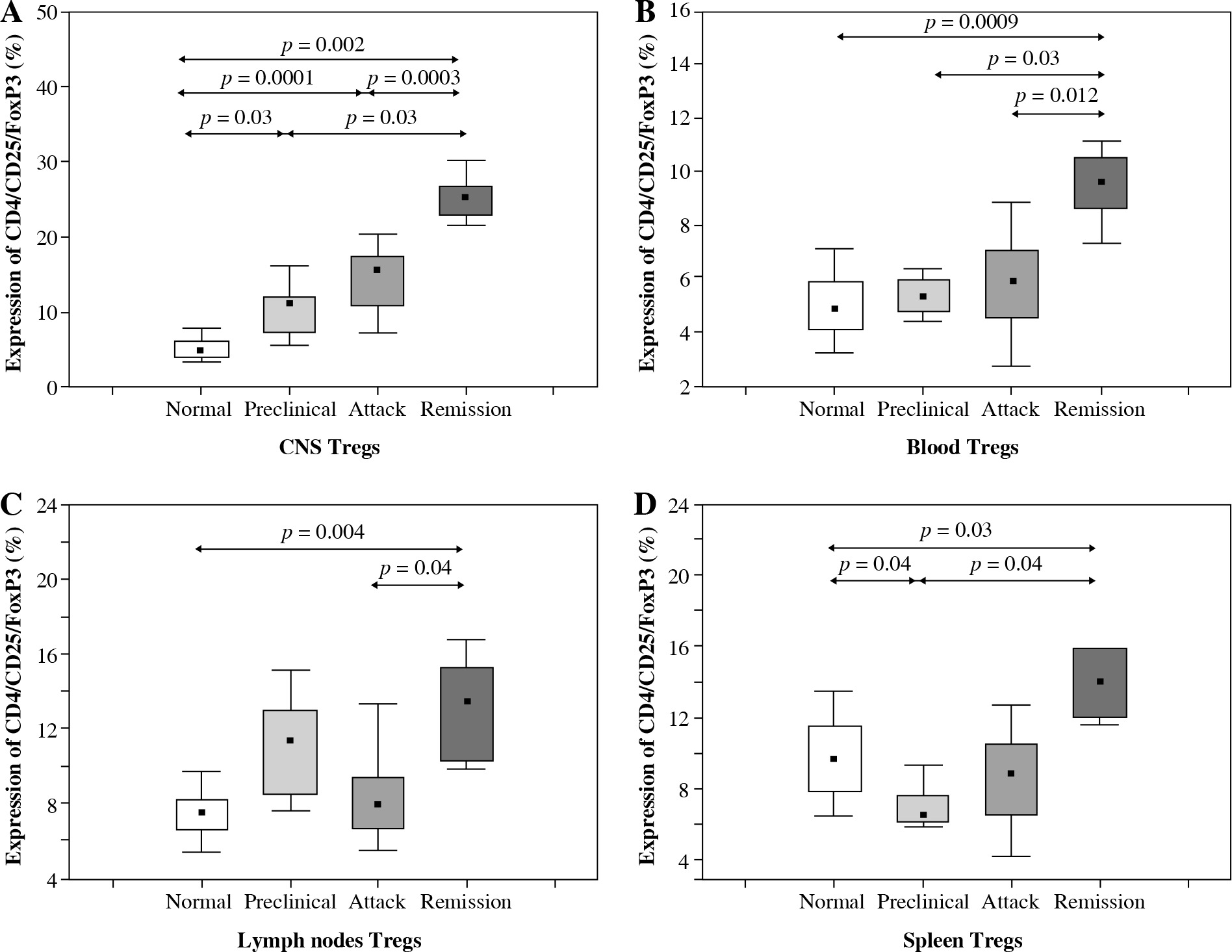
The number of Tregs circulating in the blood of control mice, animals from preclinical phase, initial attack, and remission of EAE was significantly different (p = 0.0001, Kruskal-Wallis test). Further analysis revealed that the number of Tregs was significantly higher during EAE remission than in the blood of control mice, preclinical phase, and initial attack of EAE (p = 0.0009, p = 0.03, and p = 0.012, respectively; Mann-Whitney test) (Fig. 2B).
The number of Tregs isolated from the lymph nodes was also different in groups of control mice, animals from preclinical phase, initial attack, and remission of EAE (Kruskal-Wallis test, p = 0.0002). During the remission of EAE, the number of Tregs in lymph nodes was significantly higher than in the control group and the group of initial EAE attack (p = 0.004 and p = 0.04, respectively; Mann-Whitney test) (Fig. 2C).
Similarly, there was also a significant difference in the number of Tregs detected in the spleen between the control mice, mice from preclinical phase, first attack, and remission of EAE (p = 0.0002, Kruskal-Wallis test). The highest number of Tregs was observed during the remission of EAE. Significant differences in the number of Tregs were observed between the following groups of mice: remission vs. control group, remission vs. preclinical phase, and preclinical vs. control mice (p = 0.03, p = 0.04, and p = 0.04, respectively; Mann-Whitney test) (Fig. 2D).
Immunohistochemical localization of Tregs in the CNS during EAE
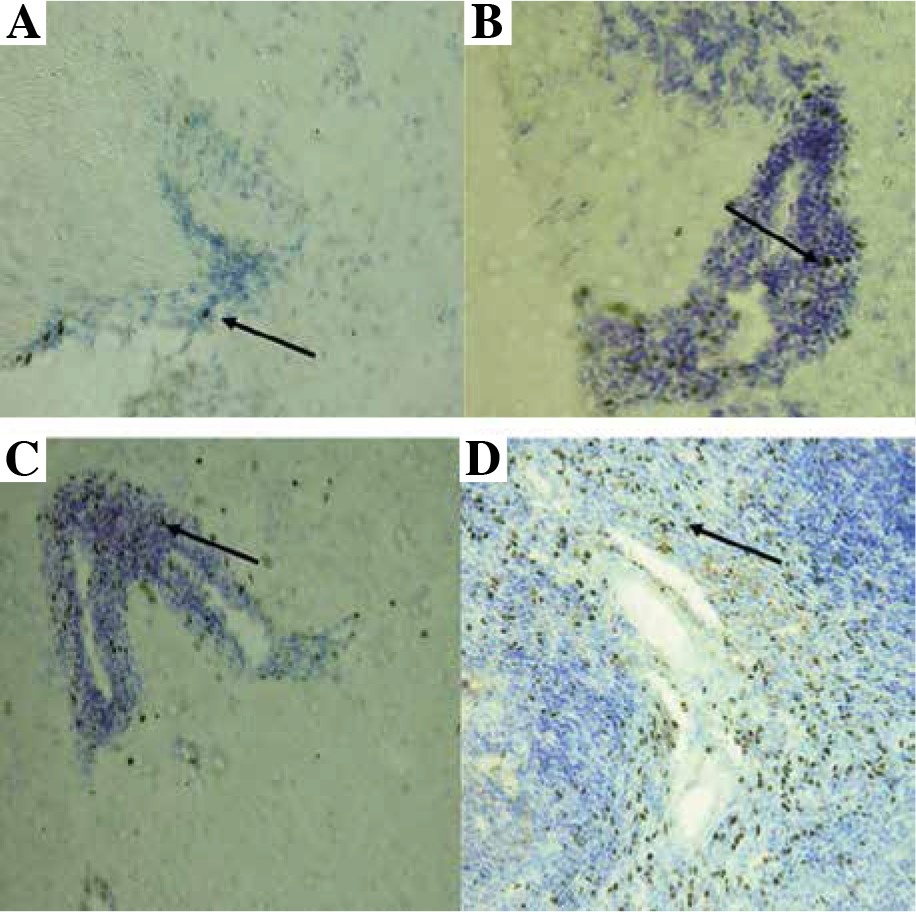
Foxp3-positive cells were detected in the CNS during different stages of EAE with immunohistochemistry. Their morphology suggested that they were lymphocytes. A minimal number of Tregs was also detected in the CNS of control unimmunized mice (Fig. 3A). Their number increased evidently during the initial attack (Fig. 3B) as well as in remission of EAE (Fig. 3C). Positive control for staining procedure were Tregs detected in spleen (Fig. 3D).
Fig. 3
Localization of Tregs (Foxp3-positive cells) (brown cells, arrows) in the brain and spleen of experimental autoimmune encephalomyelitis (EAE) mice. Staining was developed using diaminobenzidine substrate and sections were counterstained with hematoxylin and eosin. Representative brain sections are shown from: healthy control (A), initial attack (B), and remission (C) of the disease; section from the spleen (D) served as a positive control for staining (original magnification 20×)
Expression of CCR6 on Tregs from the CNS and peripheral tissues during EAE
Flow cytometry analysis of CCR6 expression on Tregs from the CNS of control mice, animals from preclinical phase, initial attack, and remission of EAE showed significant differences (p = 0.0005, Kruskal-Wallis test). Further analysis confirmed an increase of this expression on Tregs from the CNS during preclinical phase, initial attack, and remission of the disease compared to controls (p = 0.037, p = 0.037, and p = 0.023, respectively; Mann-Whitney test) (Fig. 4A). Also, CCR6 expression on Tregs was significantly higher in the initial attack rather than in preclinical phase and remission of EAE (p = 0.047 and p = 0.037, respectively; Mann-Whitney test) (Fig. 4A).
Fig. 4
CCR6 protein expression on Tregs from the central nervous system (CNS) (A), blood (B), lymph nodes (C), and spleen (D) of animals from preclinical phase, initial attack, and remission of experimental autoimmune encephalomyelitis (EAE) assessed by flow cytometry. Data is derived from a CD4+ Foxp3+ cell gate, n = 7 per group, Mann-Whitney test with Bonferroni correction
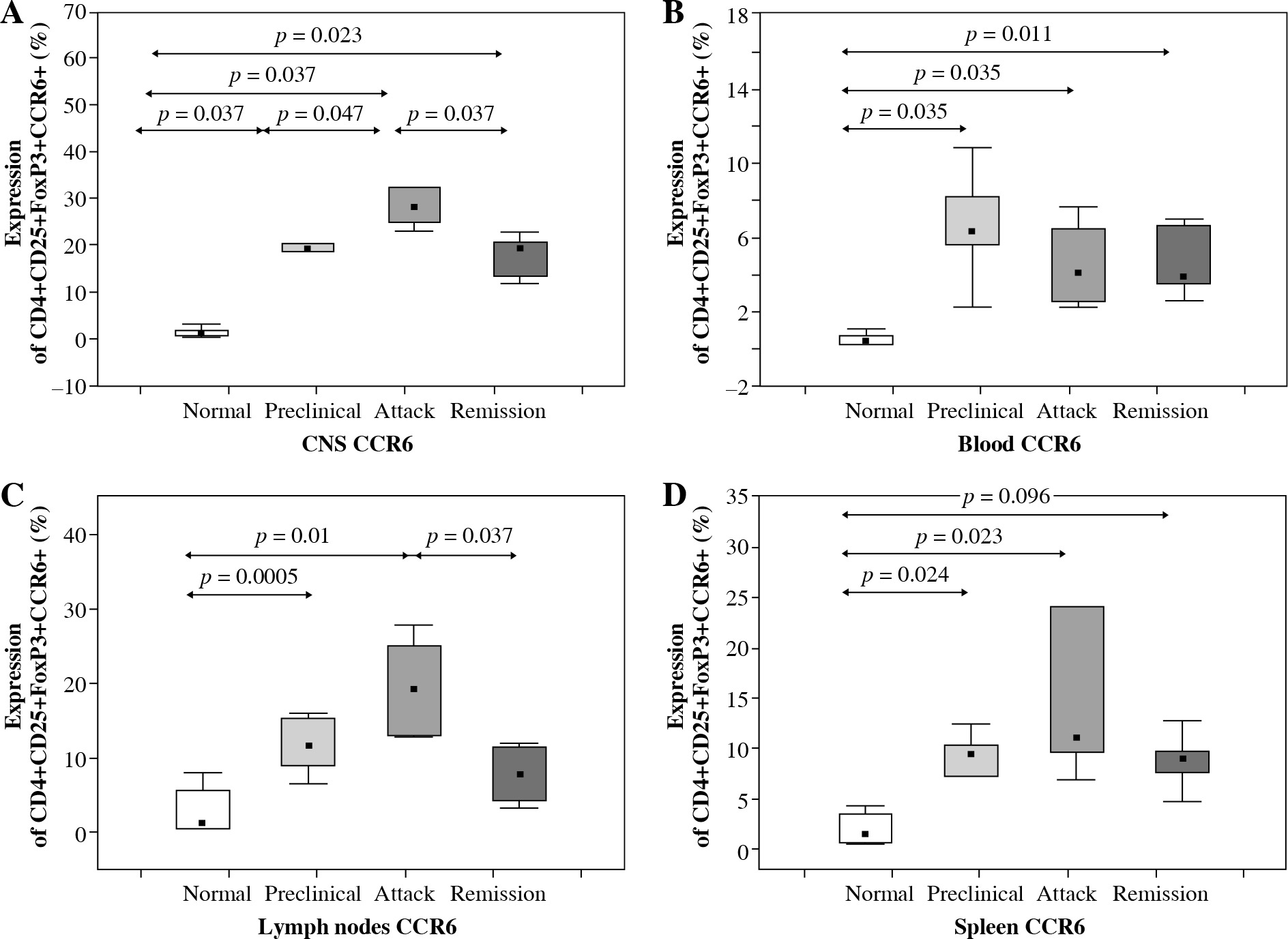
CCR6 protein expression on blood Tregs from control mice, animals from preclinical phase, initial attack, and remission of EAE was significantly different (p > 0.05, Kruskal-Wallis test). The expression of CCR6 on Tregs from the blood during preclinical phase, initial attack, and remission of EAE was significantly higher than in control unimmunized group (p = 0.0035, p = 0.0035, and p = 0.011, respectively; Mann-Whitney test) (Fig. 4B).
Additionally, the expression of CCR6 was analyzed on Tregs isolated from the lymph nodes of control unimmunized mice and animals from preclinical phase, initial attack, and remission of EAE. This analysis revealed significant difference of CCR6 expression on Tregs (p = 0.0003, Kruskal-Wallis test). CCR6 expression on Tregs from the preclinical phase and initial attack of EAE was higher than in the control group (p = 0.0005 and p = 0.01, respectively, Mann-Whitney test). Moreover, the expression of this receptor was lower during remission in comparison to the initial attack (p = 0.037, Mann-Whitney test) (Fig. 4C).
CCR6 protein expression on Tregs from the spleen of control mice, animals from preclinical phase, initial attack, and remission of EAE was significantly different (p = 0.0001, Kruskal-Wallis test). CCR6 expression on Tregs from the spleen increased during the preclinical phase, initial attack, and the remission of EAE compared to the control group (p = 0.024, p = 0.0023, and p = 0.0096, respectively; Mann-Whitney test) (Fig. 4D).
Modulation of EAE by delivery of CCR6-deficient Tregs
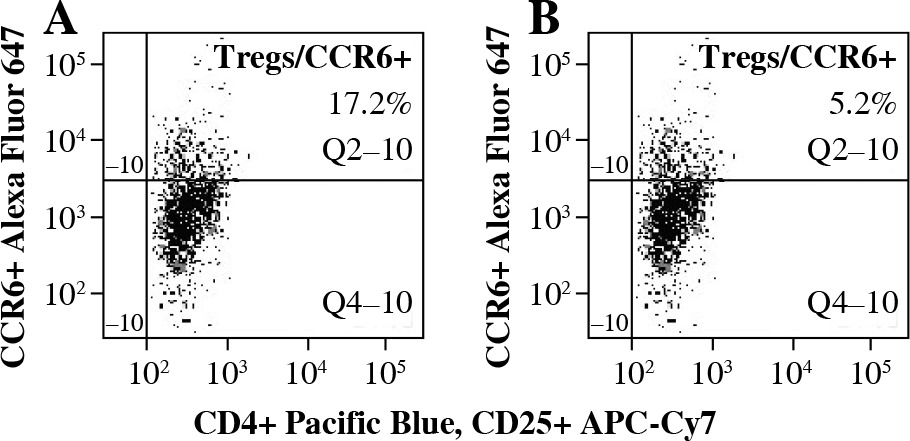
To analyze the functional effect of CCR6 deficiency on Tregs on the clinical course of EAE, Tregs with blocked CCR6 were delivered to recipient EAE mice (as described in “Material and methods” section). CCR6 protein expression on unmanipulated Tregs from lymph nodes was at the level of 17.2% (Fig. 5A) and on Tregs blocked with anti-CCR6 antibody was 5.2%, as measured by flow cytometry (Fig. 5B).
Fig. 5
Expression of CCR6 on Tregs from lymph nodes of experimental autoimmune encephalomyelitis (EAE) mice with disease remission before (A) and after (B) blockade of CCR6 with specific antibody (concentration 4.5 µg/ml) assessed by flow cytometry
The onset of EAE was seen in all three analyzed groups at the same time, but the effector phase of disease significantly changed in the group with blocked CCR6 Tregs (Fig. 6). These animals showed significantly milder disease. Statistically significant amelioration of severity of the disease was seen at days 20-23 (EAE vs. CCR6-deficient Tregs: p = 0.049, p = 0.035, p = 0.023, and p = 0.044, for consecutive days, respectively; Mann-Whitney test). During the remission phase, unmanipulated EAE have shown higher index of EAE than animals with CCR6-deficient Tregs (p = 0.035 for day 27, and p = 0.044 for day 28; Mann-Whitney test). When the second attack of the disease appeared in untreated animals, the group of EAE mice, which received Tregs with blocked CCR6, became again significantly less affected (p = 0.019 for day 32, p = 0.018 for day 33, and p = 0.013 for day 34; Mann-Whitney test). The same situation was observed in the second remission (p = 0.02 for days 37-39, Mann-Whitney test) (Fig. 6).
Fig. 6
Modulation of experimental autoimmune encephalomyelitis (EAE) clinical course by passive transfer of Tregs or Tregs with blocked CCR6. Tregs were isolated and blocked as described in “Material and methods” section. Data is expressed as the mean index of EAE for all mice in each group. Bars shows SD (EAE – unmanipulated EAE group used as morbidity control, Tregs – EAE group which received unblocked Tregs, Tregs + anti-CCR6 – EAE group which received Tregs with blocked CCR6)
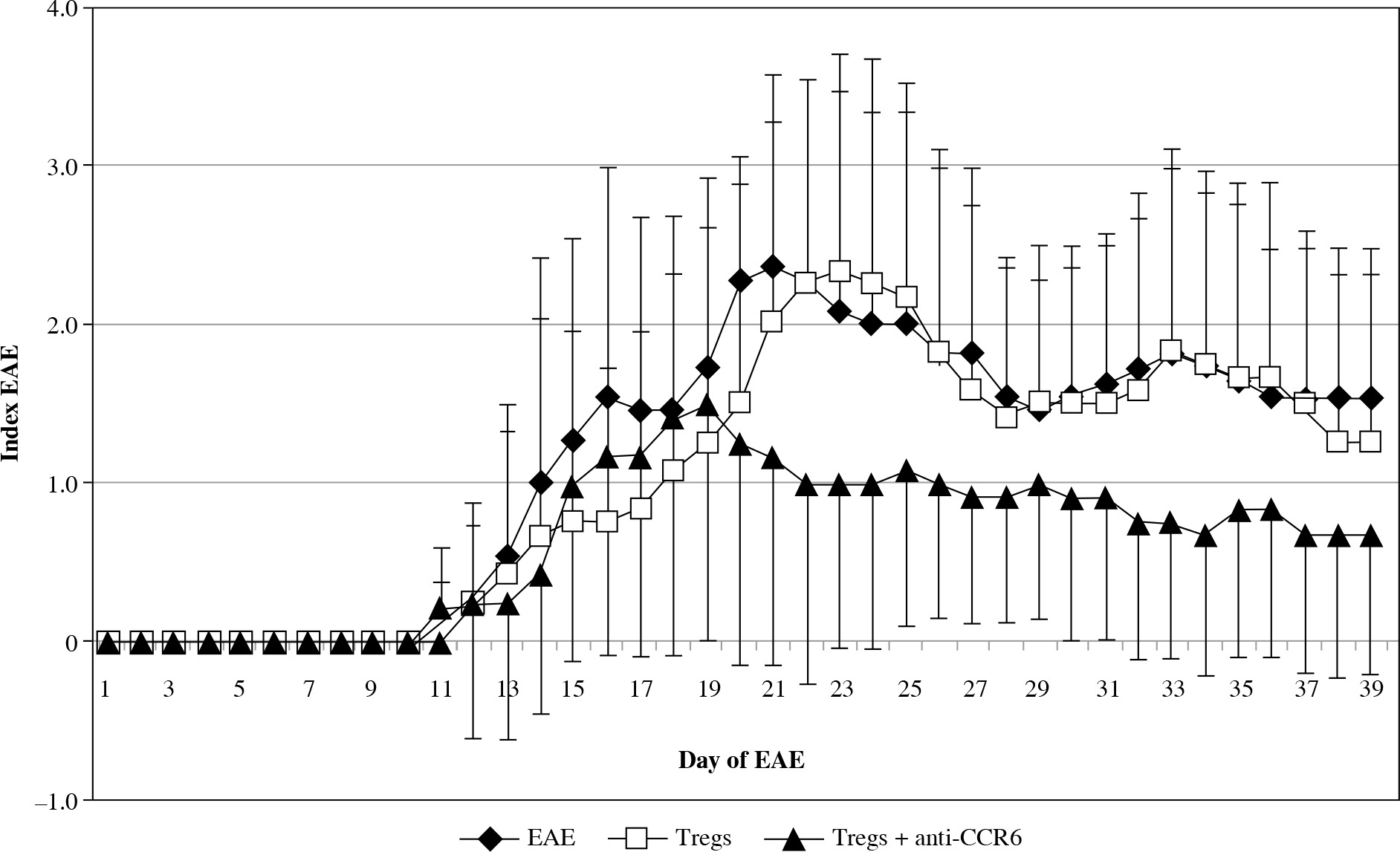
The first and second attack of EAE was almost invisible in mice that received Tregs with blocked CCR6 in comparison to mice with unmanipulated Tregs. Statistical differences in disease severity between these groups appeared at the first attack of disease (days 22-25, p = 0.023, p = 0.016, p = 0.025, and p = 0.04, respectively for consecutive days; Mann-Whitney test) and at the second attack of EAE (days 32-34, p = 0.047, p = 0.036, and p = 0.026, respectively; Mann-Whitney test). During the second remission of disease in contrast to the first one, mice with Tregs without CCR6 were again significantly less affected (p = 0.043 for day 37, Mann-Whitney test) (Fig. 6).
Discussion
Tregs are induced in periphery after targeting foreign antigens presented by dendritic cells (DCs) [32]. To travel to various tissue sites during development and inflammatory conditions, they need to regulate the expression of receptors for adhesion of molecules and chemokines [11, 33, 34]. Tregs in mice and humans express many trafficking chemokine receptors, including CCR4 [12, 35, 36], CCR5 [37], CCR7 [38], CCR8 [12], and CCR10 [39]. They can be upregulated in inflamed tissues [40-44] as well as in secondary lymphoid organs [45, 46].
In the present study, we have reported an increased accumulation of Tregs in the CNS during preclinical phase, initial attack, and remission of EAE, an experimental model of multiple sclerosis (MS). Immunohistochemical staining confirmed the increased number of Tregs during EAE in the CNS. Additionally, we demonstrated that the number of Tregs in the blood, lymph nodes, and spleen was the highest during the remission of EAE. Similar results for the CNS and lymph nodes were presented by Villares et al. [47].
There are several studies emphasizing the important role of Tregs in suppression of autoimmune diseases [48, 49]. It has been shown that amelioration of EAE is possible by transferring of Tregs from naïve mice [40, 50]. Moreover, a depletion of CD25+ T cells inhibited the recovery from EAE [43, 50, 51]. We observed that Tregs from lymph nodes of mice from EAE remission phase and transferred into recipient mice during preclinical phase of EAE did not significantly changed the course and severity of the disease. Opposite results were obtained by McGeachy et al., but they were using a different experimental protocol. In their study, lower number of unmanipulated Tregs (2 × 104 cells) was transferred much earlier, either 1 day before or 1 day after immunization of mice [43]. These authors showed that CNS-derived Tregs significantly reduced signs of EAE [43].
Here, we observed increased expression of CCR6 on Tregs during three successive phases of EAE (preclinical, initial attack, and remission). Besides elevated expression of CCR6 on Tregs from the CNS, such expression increased also on Tregs from the blood, lymph nodes, and spleen. Interestingly, the pattern of CCR6 expression on Tregs was similar in all three analyzed phases of EAE in the blood and spleen in contrast to the expression observed in the CNS and lymph nodes. In the CNS and lymph nodes, we found significantly increased expression of CCR6 on Tregs during the initial attack when compared to the remission of EAE. Kleinewietfeld et al. reported similar results for CNS [21]. Additionally, during the remission of EAE, we observed an increased number of Tregs in lymph nodes, but subpopulation of CCR6+ Tregs decreased. Probably, during remission, CCR6+ Tregs leave the lymph nodes or they downregulate CCR6 at that time.
Similarly, the expression of CCL20, a CCR6 ligand, in the CNS and lymph nodes differs during all three phases of EAE [52]. This expression increases during the preclinical phase of EAE, which later is strongly upregulated during the initial attack and returns to the baseline level during the remission of EAE [5, 52]. The highest production of CCL20 in lymph nodes of EAE mice was observed during preclinical phase of EAE [52]. Probably, CCR6+ Tregs accumulate in the CNS and lymph nodes during the preclinical phase and/or attack of EAE. Whilst the disease decreases, the level of CCL20 both in the CNS and lymph nodes diminish, which correlates with decreased number of CCR6+ Tregs in tissues. This correlation suggests that CCL20 may stimulate the migration of CCR6+ Tregs during EAE.
CCR6 plays a role in regulating the migration of T cells and B cells patrolling the normal CNS [23]. Similarly, CCR6+ T cells freshly isolated from blood of normal donors can migrate to CCL20 in vitro [53]. This observation confirms that CCR6 is essential not only in pathological but also in physiological processes of the CNS. Furthermore, the expression of CCR6 is detected on Tregs and CD45RO+ effector cells [23, 54]. It is possible that in a steady state condition (healthy organism), the major activity of Tregs is to suppress DCs, mostly their maturation [5]. Antigen-activated CD45RO+ cells are able to fully release immature DCs from suppression, which activates DCs and abrogates Tregs ability to downregulate DCs markers [5], including co-stimulatory molecules CD80/CD86 [2, 55], CCR7, and HLA-DR [54] as well as proinflammatory cytokines [55]. It is known that highly activated DCs can be resistant to suppression [5, 56]. It was reported that the interaction of CD40 on DCs with CD154 (CD40L) on CD4+CD25- T cells releases DCs from control of Tregs. This has been confirmed by blocking of CD40 on activated DCs in vitro [5].
CCL20 – the only known ligand for CCR6 [57, 58] is required for the recruitment of CD45RO+ memory T cells to the site of antigen presentation, which leads to immune cell activation [23, 53]. It was suggested that CD45RO+ cells expressing CCR6 play an important role in the early phase of inflammatory response [53]. Both CD4+ T cells [57] and immature dendritic cells [59] express CCR6 and traffic to the lymph nodes towards CCL20, which is expressed by endothelial cells upon activation by TNF alpha [60]. TNF alpha is important for innate immunity and is expressed in lymph nodes [60, 61]. Reboldi et al. have shown that CCL20 can be constitutively expressed by endothelial cells from the choroid plexus in CNS ventricles [23].
Recently, it was reported that immunocompetent cells that enter the CNS very early during inflammatory response (the first wave) are autoreactive Th17 cells expressing CCR6, which lead to a second wave of migration of antigen non-specific T cells to the CNS [23]. The first influx of CCR6+ Th17 cells is needed for cellular activation within inflammatory foci, and leads to a production of inflammatory cytokines and chemokines, which in turn activate the blood-brain barrier (BBB) and initiate the influx of large number of inflammatory effector cells [23]. It was also confirmed that Th17 cells, but not Th1 and Th2 T cells, highly express CCR6 [22]. CCR6 may be essential in the priming phase of EAE and is not involved in the effector phase of EAE [62]. It has been reported that administration of anti-CCR6 [62] and anti-CCL20 [52] antibodies before passive transfer of PLP 139-151 reactive lymphocytes had not changed the course of EAE. Therefore, it was suggested that CCR6 is not involved in the effector phase of EAE [62]. Additionally, the lack of CCR6 on Th17 cells impaired the recruitment of those cells to the CNS during EAE [22]. Moreover, neutralization of IL-17 strongly inhibits EAE [24, 27]. Those observations confirmed the essential role of Th17 cells in the induction of EAE and the important function of CCR6 in migration of those cells to inflamed tissues.
In this study, we investigated whether CCR6 on Tregs from EAE animals given to recipients has an impact on disease’s course and severity. We showed that Tregs with blocked CCR6 may decrease acute EAE severity in recipients. This group of mice developed significantly milder neurological damage during the first and the second attack of disease compared to the control EAE group. It seems that CCR6 on Tregs is needed for their migration to the CNS, and its absence significantly reduce the signs of EAE.
Villares et al. showed that CCR6+/+ Tregs transferred to CCR6-/- mice before EAE induction reduced the severity of EAE. They concluded that CCR6 is required for Tregs to downregulate EAE [47]. However, the results of this study are very difficult to compare with ours because of different experimental protocols. Firstly, they transferred CCR6+/+ Tregs much earlier, even before EAE induction. We delivered Tregs with blocked CCR6 to recipients on days 7-8 after EAE induction during preclinical phase of the disease. Secondly, we isolated Tregs from EAE mice during remission, whereas Villares et al. used normal (unimmunized) animals [47]. Moreover, the recipient animals in their study were CCR6-/- animals, whereas we used wild type mice. Our EAE models were also different. They used MOG-induced model in C57Bl/6 mice, while our model was PLP-induced in (SWRxSJL)F1 mice. Additionally, our Tregs were collected from draining lymph nodes, whereas Villares et al. collected Tregs from spleen [47]. All above-mentioned differences in study design may justify different conclusions from our experiments. However, the results from both studies taken together may indicate the important, but complex role of CCR6 on Tregs in EAE pathogenesis. Further studies are needed to clarify these observations.
In our study, we demonstrated that Tregs increasingly migrate to the CNS during subsequent phases of EAE (preclinical, initial attack, and remission). Moreover, we observed an increased expression of CCR6 on Tregs in the CNS, blood, lymph nodes, and spleen in all phases of EAE. To further analyze the functional significance of such expression in EAE pathology, we modulated the clinical course of this MS model using Tregs with blocked CCR6. EAE mice, which received CCR6-deficient Tregs, showed significant amelioration of the disease severity. This observation suggests that CCR6 on Tregs may be a potential target for the future therapeutic interventions in MS.