
Introduction
Autoimmune bullous diseases (ABDs) featuring autoimmunity to structural proteins are a heterogeneous, relatively rare group of potentially life-threatening mucocutaneous dermatoses, defined by autoantibodies against adhesion proteins in the stratified squamous epithelium or its basement membrane zone (BMZ), which in the case of the epidermis is known as the dermoepidermal junction (DEJ). Clinically, ABDs manifest with cutaneous and/or mucosal blisters and their evolutionary lesions [1], but patients quite frequently present unobvious, less textbook or overlapping clinical signs which can cause delays in diagnosis. Depending on the level of blister formation, ABDs are classified into intraepithelial illnesses known as pemphigus diseases, and subepithelial diseases [2]. The intertwined genetic factors, autoimmune responses, and mediators of inflammation are involved in pathogeneses of these dermatoses [3]. ABDs require clear-cut diagnosis as usually aggressive immunosuppression, having serious side-effects, is needed. Nowadays, the diagnosis relies on a combination of various criteria: clinical features as well as the detection of skin/mucous membrane-bound and circulating autoantibodies. Autoimmunity can be detected using tissue imaging, and serum biochemical-molecular methods, among which the enzyme-linked immunosorbent assay (ELISA) is currently widely available [4, 5]. Immunofluorescent techniques were introduced for diagnosing ABDs in the early 1960s [6].
Despite various advances in serological diagnosis, direct immunofluorescence (DIF) microscopy still remains most widely used and is regarded as the diagnostic gold standard for ABDs [7, 8]. DIF microscopy is a procedure that demonstrates the antibodies bound in vivo to antigens in the skin or mucosae. With DIF, intercellular intra-epidermal/epithelial staining of immunoreactants is seen in pemphigus diseases, while subepithelial ABDs, excluding dermatitis herpetiformis, are characterized by linear staining along the BMZ/DEJ [7, 9]. A 3-4 mm punch biopsy is optimal for DIF study; the perilesional uninvolved skin site is crucial. Interpretation of DIF examination is based on key features: types of immunoreactants deposited (immunoglobulin (Ig) G, IgG1, IgG4, IgA, IgM and C3 are separately evaluated with monospecific fluorescein-conjugated antibodies in our tertiary referral centre), the site and patterning of immunoreactants deposition, and the intensity of a fluorescence signal usually assessed using subjective grading scales [2]. Automated systems utilizing artificial intelligence approaches are needed for evaluating DIF images in ABDs. DIF microscopy was reported to have sensitivity in the range of 82-91% and specificity of 98% [10, 11].
It is known that IgG is the most abundant isotype in the human serum, constituting about 80% of the total serum immunoglobulin [12]. There are four IgG subclasses in humans numbered 1 to 4 in the order of their discovery and serum concentration [13]. IgG1, the most abundant subclass, is present in serum in the range 5-11 mg/ml, whereas IgG4, the least abundant one, is present in concentration of 0.35-0.51 mg/ml [13, 14]. IgG4 is a very odd and dynamic antibody. Regulation of IgG4 production is dependent on help by T-helper type 2 (Th2) cells and the view is that this antibody appears only after prolonged immunization [15]. In most diseases IgG4 antibodies are innocent bystanders. In general, IgG4 antibodies have poor complement- and leucocyte-activating capacity, but in some situations IgG4 antibodies have an association with pathology, e.g. in autoimmune pancreatitis, the sclerosis-associated hyper-IgG4 syndrome and subepithelial ABDs. The mechanism for the association of IgG4 with these diseases is not fully understood; further studies may prove their pathogenic role and unravel its scope in those conditions. IgG4 antibodies may predominate in pemphigus diseases, suggesting that the intraepithelial blistering process does not depend on complement activation [15-17]. Purified IgG4 autoantibodies were shown to induce dermal-epidermal separation in an ex vivo skin model [18]. IgG1 antibodies mediate tissue damage, whereas IgG4 antibodies seem to mainly mediate acantholysis [16]. Disease-specific subclass distribution may be responsible for false-negative results in diagnostics of ABDs. Bowszyc-Dmochowska and Dmochowski proposed to routinely use anti-IgG4 conjugates for this diagnostic purpose [19]. Dmochowski et al. in the 1990s detected the IgG4-mediated autoimmunity in pemphigus diseases with indirect immunofluorescence (IIF) on tissue sections and transfected culture cells as well as immunoblotting, suggesting that the IgG4-mediated pemphigus type immune response is a true finding not depending on the particular laboratory method used [20, 21]. Therefore, evaluating the IgG4 autoimmune response can be implemented for diagnosing pemphigus diseases and differentiating them within the spectrum of ABDs and from other illnesses of various etiopathogeneses showing similar clinical features. Accordingly, Pietkiewicz et al., assessing serum IgG4 antibody, concluded that this approach significantly increases autoimmunity detection in IgG-mediated subepithelial ABDs initially diagnosed/screened with IIF [22]. Our patient with a relapsing classic/mechanobullous variety of epidermolysis bullosa acquisita (EBA) requiring tracheostomy despite treatment with numerous traditional therapies and rituximab still had active mucocutaneous lesions 3 months after its introduction. In the IIF mosaic assay, the patient at that stage had negative IgG, but positive IgG4 EBA-type antibodies, showing the usefulness of IgG4 antibody evaluation [23].
In this study, independently from pathogenetic considerations, we aimed at streamlining the diagnostic process of ABDs by developing an innovative DIF procedure with fluorescein conjugate against IgG + IgG4 that had been used before just for IIF [24].
Aim of the study
The aim of this study was to compare the diagnostic accuracy of traditional DIF (DIFt; separate IgG, IgG1, IgG4, IgA, IgM and C3 deposits detection) and modified DIF (DIFm; simultaneous IgG + IgG4 deposits detection instead of separate IgG and IgG4 deposits detection) in routine diagnostics of IgG-mediated ABDs.
Material and methods
Patients
The study was conducted in the setting of a tertiary referral centre for autoimmune blistering diseases of a Central European university dermatology department.
A selection of 18 Slavic patients (3 males and 15 females) with IgG-mediated ABDs was examined. The study group consisted of 7 patients with pemphigus dermatoses, namely 4 with pemphigus foliaceus (PF) and 3 with pemphigus vulgaris (PV), and 11 with subepithelial ABDs, namely 7 with bullous pemphigoid (BP), 1 with Brunsting-Perry pemphigoid, 2 with mucous membrane pemphigoid (MMP) and 1 with pemphigoid gestationis (PG). The age of the patients with pemphigus ranged from 67 years to 83. The mean age was 76.28 years. The age of the patients with BP ranged from 66 years to 90, and the mean age was 80.88 years.
The diagnoses were established based on the following criteria: 1) clinical features – mucocutaneous blisters and their evolutionary lesions on predilection sites and any other signs and symptoms suggesting ABDs, 2) appropriate DIF patterns, 3) appropriate results of serum studies with biochemical-molecular multianalyte ELISA containing 6 antigens (DSG1, DSG3, BP180, BP230, envoplakin, type VII collagen) (Euroimmun, Lübeck, Germany).
All patients had no treatments for ABDs before biopsy taking for DIF.
Direct immunofluorescence procedures
Traditional DIF of perilesional skin deposits was performed following the procedure previously described [3]. The tissue sections were incubated in a humid chamber for 30 minutes at room temperature (RT) with commercially available fluorescein isothiocyanate (FITC)-conjugated anti-human IgA, IgM, IgG, and C3 rabbit polyclonal antibodies (Dako, Denmark) and FITC-conjugated anti-human IgG subclasses: IgG1 and IgG4 murine monoclonal antibodies (Sigma, USA). DIFt with the separate assessment of IgG, IgG1, IgG4 deposits was modified in the procedure of DIFm to enable simultaneous assessment of IgG + IgG4 deposits using an IgG4 enriched antitotal IgG FITC conjugate (Euroimmun, Germany) aiming at replacing the traditionally used separate anti-IgG and anti-IgG4 FITC conjugates. All FITC conjugates were used at a working dilution of 1 : 100 in phosphate buffer saline (PBS). The samples were then washed in PBS (pH 7.2) at RT for 15 minutes with gentle agitation. Then, slides were coverslipped and examined.
In order to limit subjectivity, the assessment of DIF specimens for unequivocal identification of the types of immunoreactants present was performed by two independent evaluators, using two different fluorescence microscopic systems: blue light-emitting diode technology-operated microscopy (EuroStar III Plus microscope, Euroimmun, Germany) (concluded to be preferable for routine laboratory diagnostics of ABDs) and short arc mercury lamp-operated microscopy (BX40, Olympus, Japan) at identical objective magnifications (20×, 40×) [25].
Results
The detailed results of DIFt and DIFm are shown in Table 1, whereas DIFm and DIFt findings in representative patients are shown in Figure 1.
Fig. 1
Representative autoimmune bullous disease (ABD) patients. A middle-aged man with a relapse of pemphigus foliaceus (PF2) presenting an asymptomatic erythema annulare-like lesion on the medial surface of the thigh (A, at the top), in whom an elevated level (value 5.97, cut-off ratio = 1.0) of serum immunoglobulin (Ig) G antibodies to DSG1 was found with the multianalyte ELISA, having IgG4 (+++) pemphigus deposits (A, in the middle) detected with traditional direct immunofluorescence (DIFt), and IgG + IgG4 (+++) deposits (A, at the bottom) detected with DIFm. An elderly woman with bullous pemphigoid (BP2) presenting itchy wheal-like lesions and blood-filled long-lasting blisters/ vesicles on the medial surface of the thigh (B, at the top), in whom an elevated level of serum IgG antibodies to BP180 (value 6.33, cut-off ratio = 1.0) and BP230 (value 1.6, cut-off ratio 1.0) was found with the multianalyte ELISA, having IgG4 (++) (B, in the middle) and C3 (+) linear deposits along the DEJ detected with DIFt, and IgG + IgG4 (++) (B, at the bottom) linear deposits along the DEJ detected with modified direct immunofluorescence (DIFm). All DIF images shown were visualized with the blue light-emitting diode technology-operated microscopy system (original objective magnifications 40×)
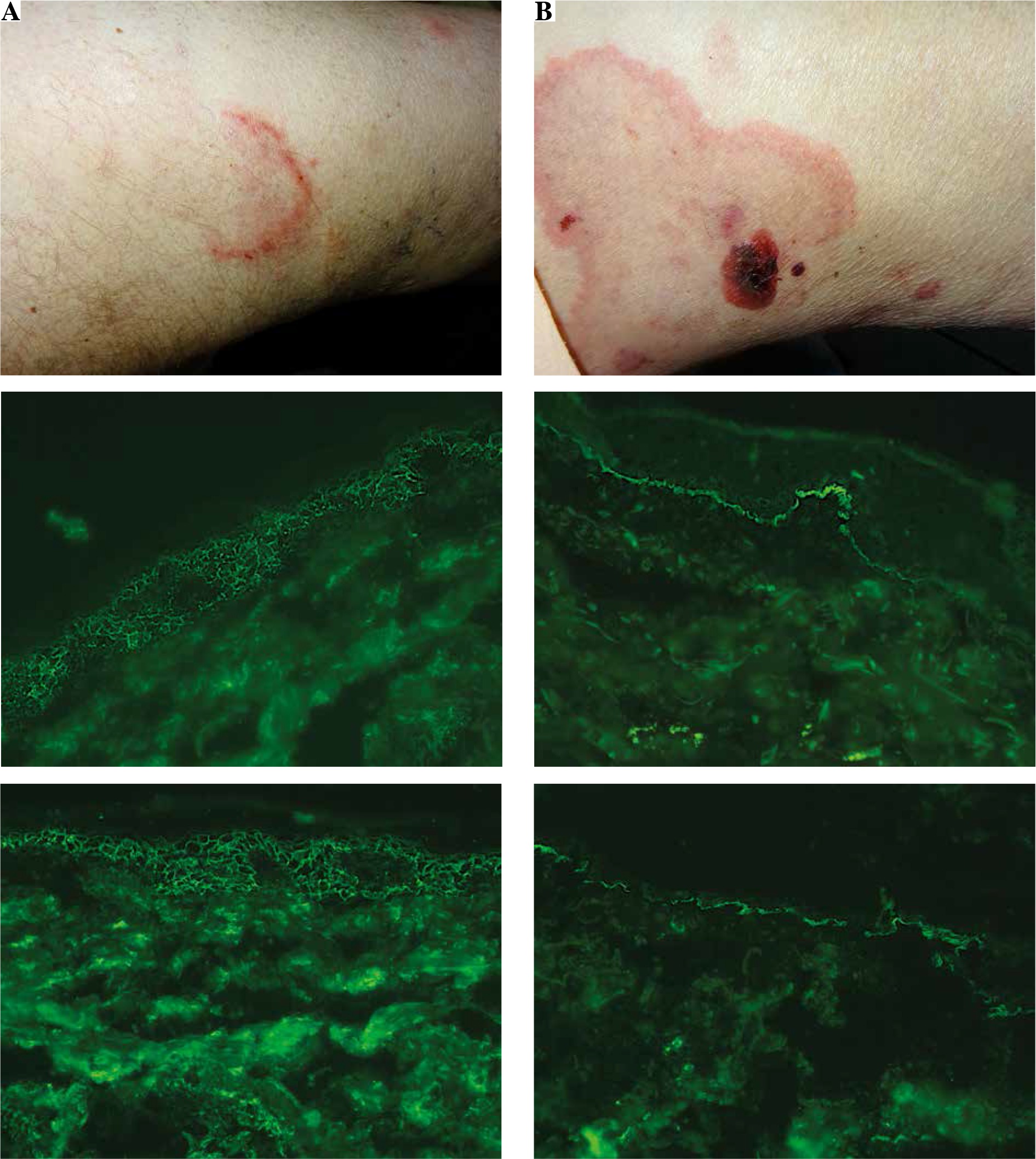
Table 1
Detailed results of DIFt and DIFm. IgG, IgG1, IgG4, IgA, IgM and C3 (DIFt) and IgG + IgG4 (DIFm) deposits were evaluated in all patients. Undetected deposits of evaluated immunoreactants in DIFt were not mentioned
[i] PF – pemphigus vulgaris, PV – pemphigus foliaceus, BP – bullous pemphigoid, MMP – mucous membrane pemphigoid, B-P pemphigoid – Brunsting-Perry pemphigoid, PG – pemphigoid gestationis, DIF – direct immunofluorescence, DIFt – traditional DIF, DIFm – modified DIF, P-gus – pemphigus “chicken wire” or, in the case of IgG4, “dew drops on spider web” patterns of immunoreactants deposits, DEJ – linear pattern of immunoreactants deposits along the dermal-epidermal junction, “–” – immunoreactant deposit not detected, “+/–” – immunoreactant deposit of borderline intensity, “+” – immunoreactant deposit of weak intensity, “++” – immunoreactant deposit of intermediate intensity, “+++” – immunoreactant deposit of strong intensity
The statistical analysis using Fisher’s exact test revealed a statistically significant relationship (p = 0.0186) between DIFm and DIFt results (Table 2).
Table 2
Analysis of positive DIF results (number of patients with detected IgG immunoreactants deposits) and negative DIF results (number of patients with undetected IgG immunoreactants deposits)
| DIFt positive | DIFt negative | Statistical analysis | |
|---|---|---|---|
| DIFm positive | 13 | 1 | Fisher’s exact test p = 0.0186 |
| DIFm negative | 1 | 3 |
The agreement of detectability of IgG immunoreactants deposits, namely, in our study any of IgG, IgG1, IgG4 and IgG + IgG4, was obtained in 16 of 18 ABD cases (88.89%), as positive results in both DIFt and DIFm were obtained in 13 cases and negative results as exclusively C3 deposits with DIFt found in both DIFt and DIFm were obtained in 3 cases. One ABD case (Brunsting-Perry pemphigoid) (5.56%) was negative in DIFm with a positive DIFt result (IgG1 deposits). One ABD case (BP) (5.56%) had only C3 deposits, but not IgG immunoreactants, in DIFt with a positive DIFm reading (IgG + IgG4 deposits).
All our patients with active pemphigus were positive in both DIFt and DIFm having deposits of IgG immunoreactants detected. Three subepithelial ABDs patients had no IgG immunoreactants detected in both DIFt and DIFm having only C3 deposits found with DIFt (2 cases of BP and 1 of PG).
Discussion
A recent update on DIF concluded that DIF still is an indispensable practical tool for diagnosing ABDs [26]. In this study we present a comparison of two methods of DIF (DIFt and DIFm) in diagnosing and differentiating ABDs. Our study was designed to assess just the detectability of immunoreactants, but not compare their intensity. The rationale for such a design is that the assessment of the intensity of immunodeposits visualized with imaging microscopic techniques always has a certain level of subjectivity regardless of unautomated grading methods used and, according to our clinical-laboratory experience, the intensity of deposits, being considerably dependent on spatial-temporal phenomena, in DIF of ABDs is the least valuable information for practicing clinicians. Nonetheless, the intensity of immunoreactant deposits evaluated using a subjective grading scale is presented in Table 1 just to detail our DIF results. To date, to the best of our knowledge, no previous study investigating the usefulness of an IgG4 enriched antitotal IgG FITC conjugate (intended for IIF study) for DIF assay has been published. An IgG4 enriched antitotal IgG FITC conjugate was used by Goletz et al. in an IIF mosaic assay with three fragments of laminin 332 in MMP [24]. The sensitivity of detecting antibodies to this heterotrimer increased when an anti-IgG4 enriched antitotal IgG conjugate was applied.
Bullous pemphigoid and pemphigus diseases are well-characterized entities within the ABD spectrum, in which the autoimmune response is heterogeneous indeed, but shows some shared peculiarities in subclass distribution. Numerous studies have clearly demonstrated that antibodies in pemphigus patients mainly belong to the IgG1 and/or IgG4 subclasses [27]. IgG4 antibodies predominate in both active PF and active PV [12, 21, 27, 28]. In contrast, in paraneoplastic pemphigus (PNP) the autoantibodies mainly belong to IgG1 and IgG2 subclasses [2, 13, 29]. The analysis of the subclass distribution of IgG antibodies in the skin of patients with BP by DIF microscopy revealed IgG4 as being the prevailing subclass, followed by IgG1 autoantibodies, while IgG2 and IgG3 autoantibodies were found only occasionally [27, 30]. Autoantibodies in MMP mainly belong to the IgG4 and IgG1 subclasses. In MMP with autoimmunity to laminin 332 autoantibodies almost exclusively belong to the IgG4 subclass [31]. In contrast to BP, in PG autoantibodies seem to belong mainly to the IgG1 and IgG3 subclasses, but not the IgG4 subclass [27].
Our results indicated that the use of DIFm seems justified in particular in the active phase of ABDs since the predominance of the IgG4 subclass is observed during the active stage of ABDs, while autoantibodies of the Th1-dependent IgG1 subclass are seen during the chronic course of these disorders [10, 16, 22]. Indeed, our previous study using DIF revealed that antibodies deposited in tissue in pemphigus and BP at their active stages belong predominantly to the IgG4 isotype [16].
It seems that the innovative DIFm method slightly increases the detectability of ABD type IgG immunoreactant deposits in relation to DIFt. An explanation for the fact that there are ABD patients in whom the deposits of IgG cannot be detected with DIF, but IgG4 are detectable, might be that that FITC-anti-IgG conjugates are produced using normal sera from healthy individuals and in such sera IgG4 concentration is low, so the sensitivity of such a conjugate may be too low for detection of IgG4 in a disease state.
It should be stressed that the separate evaluation of IgG1 deposits should be continued, in addition to IgG + IgG4 deposits, as IgG1 deposits can be detected, instead of IgG4 deposits, in certain cases of PG, which should be regarded as a valuable laboratory clue in the differential diagnosis of PG at the tissue level [32]. Findings obtained by Kelly et al. in PG revealed that IgG1 was the major IgG antibody subclass in both serum and tissue, being detected in sera of all PG patients studied [33], which is important as far as pathogenicity is concerned since IgG1, being the robust complement activating subclass via the classical pathway, should be regarded as the key factor in the chain of events leading to complement-dependent PG tissue damage. A Brazilian small-series study reported that the majority of patients with PG (85.7%) exhibited C3 or both C3 and IgG deposition along the DEJ in DIF [34]. Our laboratory experience with DIF is that there are certain PG sufferers in whom only C3 deposits can be detected, but not IgG immunoreactants, which is in line with the Brazilian findings that in 4 of 7 PG patients studied DIF staining revealed exclusively linear C3 deposition along the DEJ [34]. The study of Hallaji et al. on PG revealed linear deposition of C3 along the DEJ in DIF in 100% of patients with PG and linear IgG deposition along the DEJ in only 70% of patients. The linear fine IgM deposition was also noted along the DEJ in 17.4% of patients [35]. Moreover, in our study, one person diagnosed with BP after evaluating all clinical, imaging and biochemical-molecular data available had only IgG1 deposits in DIFt, without IgG + IgG4 deposits in DIFm. Thus, performing exclusive evaluation of IgG + IgG4 deposits in tissue could lead to misdiagnosis.
DIF assay of pemphigus diseases and BP often shows C3 deposits, suggesting complement activation in situ. However, this finding is not sufficient to support its significant involvement in pathogenesis of blister formation in pemphigus diseases [36]. Complement protein C3 plays an important role in activation of the complement system in classical, lectin and alternative pathways [13, 37]. In animal models using complement-deficient mice, different complement pathways are involved in the pathogenesis of different ABDs [37]. Surprisingly, only a few detailed studies have been published so far concerning the value of C3 deposits in the diagnosis of ABDs [38, 39]. Observations of Krasny et al. indicate that the finding of intercellular C3 deposits in the case of absence of IgG in normal skin is not a sign of pemphigus but, rather, a sign either of a type of drug reaction or, possibly, of some connective tissue disease [40]. It is well accepted that the mechanism of blister formation in pemphigus is independent of granulocytes or complement activation in the epidermis [36]. In contrast, the view is widely held by clinicians that the blister formation in BP depends on granulocytes and the activation of complement is necessary in this process [36]. It is true that in DIF studies of BP linear deposition of C3 at the DEJ is usually prominent. A large study of Romeijn et al. demonstrated that 250 out of 301 patients (83%) with BP had C3 depositions along the DEJ in DIF microscopy, but 13% of BP cases were negative for C3 in the DIF assay [41]. These results apparently indicate that 17% of patients may develop blisters mainly in a complement-independent manner. Furthermore, the passive transfer of IgG autoantibodies from BP patients induced blister formation in neonatal C3-deficient BP180/COL17-humanized mice without complement activation [42].
Three of our patients had only C3 deposits in the DIFt assay without deposits of IgG immunoreactants in both DIFt and DIFm. The detection of exclusively C3 deposition should not be regarded as a marker of autoimmune processes, as C3 can be activated via the lectin pathway without the participation of IgG immunoreactants. In the Białynicki-Birula et al. study, deposits of C3c along the DEJ were found in 49 of 61 (80.3%) BP patients examined, whereas C1q deposits, which are indicators of classical pathway activation, occurred only in 39 (63.9%) patients [43]. Thus, that study on complement cascade proteins indirectly indicated that the alternative pathway has a role in BP pathogenesis. It still remains to be investigated what the role of the complement system in the complex pathogenesis of ABDs actually is.
The statistical analysis of our results indicates that both DIFm and DIFt are useful methods to detect deposition of IgG immunoreactants in ABDs as a satisfactory level of agreement of DIFt and DIFm results was found. The introduction of DIFm into routine laboratory diagnostics of ABDs seems to be justified, as it enables the abandonment of separate conjugates for IgG and IgG4, which is important for cost-effectiveness as the procedure of DIFm is less laborious, requiring fewer volumes of reagents.
In conclusion, it is suggested that DIF for diagnosing and differentiating ABDs should be performed using fluorescein conjugates to IgG + IgG4, IgG1, IgA, IgM and C3.