

Introduction
Primary immunodeficiency diseases (PIDs) have increased in number over time and are now associated with about 300 genes [1]. Not all of the PIDs may appear along with obvious infectious diseases. Autoimmunity, immune dysregulation, and cytopaenia are the other clinical manifestations of PIDs [2]. Lipopolysaccharide-responsive beige-like anchor protein (LRBA), one of eight proteins that have a BEACH area [3], is a lysosomal trafficking regulator that conducts duties related to vesicular transport, autophagia, apoptosis, membrane dynamics, and receptor transmission. LRBA deficiency is one of the combined immunodeficiency syndromes with Mendelian transmission, which occurs due to LRBA defects [1]. The main features of LRBA deficiency consist of autoimmunity and immunodeficiency symptoms [4, 5]. The most reported clinical complications were autoimmune haemolytic anaemia (AIHA), immune thrombocytopaenic purpura (ITP), organomegaly, recurrent infections due to hypogammaglobulinaemia, and chronic diarrhoea [4, 5].
Defects in LRBA (OMIM number: 606453) in B cells cause a decrease in autophagia, diminishing its cytoprotective effect, thereby increasing cellular vulnerability to apoptosis and promoting the subsequent development of qualitative and quantitative B cell deficiencies [6, 7]. Defects in LRBA in T cells also decrease autophagia and increase apoptosis, with a resulting development of T cell dysregulation and autoimmune symptoms [8]. All lymphocyte subgroups may be affected at different rates. The most reported lymphocyte subsets abnormalities were reduced plasmoblast, switched memory B cells, total B cells, and Tregs. But these can also be normal [4].
The aim of this article is to report a boy with genetically confirmed LRBA deficiency, who had multiple gallstones, which have not been previously reported in LRBA-deficient patients.
Case report
An 11-year-old male patient was referred to an immunology and allergy clinic for frequent respiratory tract infections. His medical history showed that he had been diagnosed with chronic ITP since the age of six. He was diagnosed with Evans syndrome due to the occurrence of AIHA. He had received several treatments of pulse steroid, platelet suspension supplementation, and intravenous immunoglobulin (IVIG). In addition, he had received erythrocyte suspension supplementation two times. The prenatal and perinatal history was not remarkable. His parents’ marriage was not consanguineous. Physical examination revealed that the patient had a body weight of 30 kg (10th-25th centile), a height of 137 cm (10th-25th centile), ecchymotic areas on his skin, and hepatosplenomegaly. There was no BCG scar or lymphadenopathy.
Laboratory investigations showed the following findings: WBC: 10440/mm3; lymphocytes: 2590/mm3; neutrophils: 6760/mm3; haemoglobulin: 11.3 g/dl; platelets: 269,000/mm3; reticulocytes: 1.46 %; haptoglobin: 138 mg/dl (30-200); AST: 32 U/l (8-46); ALT: 35 U/l (0-40); GGT: 13 U/l (8-61); LDH: 266 U/l (95-500); and total bilirubin: 0.31 mg/dl (0.1-1.5). Immunologic investigations showed the following findings: IgG: 570 mg/dl (842-1943); IgA: 7 mg/dl (67-433); IgM: 27 mg/dl (54-392); IgE: 18.3 IU/ml; anti-A and anti-B antibody 1/32; positive direct Coombs test; C3: 0.8 g/l (0.8-2.0); and C4 too low to measure. The tuberculin skin test was anergic. His vaccine response was not checked because he had undergone IVIG treatment. It was learned that the patient had direct Coombs positivity and low C4 all the time. Bone marrow examination was normal. The EBV-DNA, CMV-DNA PCR, and anti-HIV were negative. Autoimmune lymphoproliferative syndrome (ALPS) was considered according to physical and laboratory findings. Double-negative T cells (DNTs) measured twice were 4.4% and 5.8%, respectively. He was diagnosed with ALPS based on an increase in DNTs.
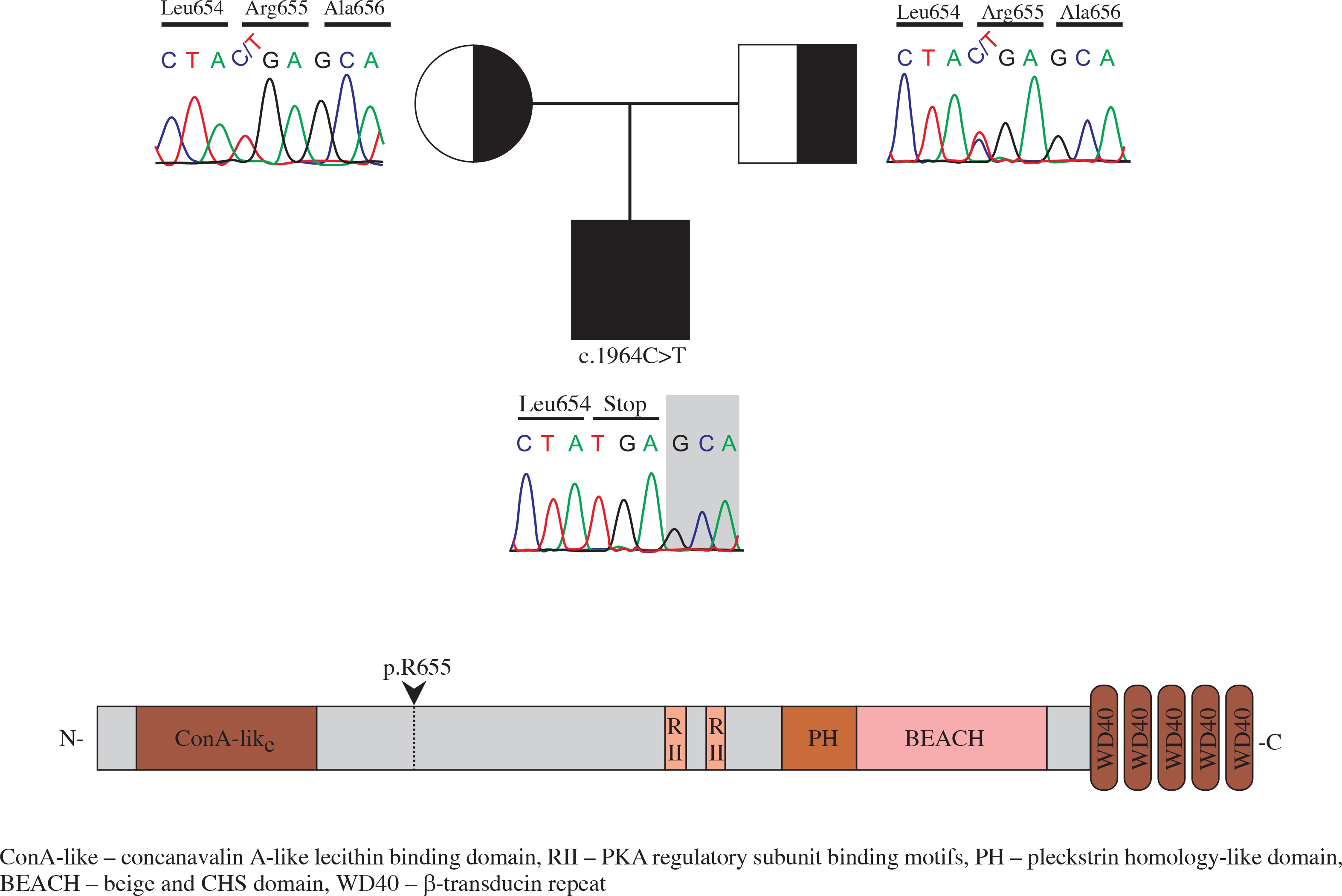
Flow cytometry analysis showed decreased memory and switched B cells, and an inverted CD4/CD8 ratio (Table 1). Common variable immunodeficiency (CVID) was thought to accompany ALPS. Regular IVIG therapy and cotrimoxazole prophylaxis were started. LRBA deficiency was considered due to CVID-ALPS overlap syndrome. Homozygous mapping was performed, followed by exome sequencing. A homozygote mutation (c.1964C>T) in LRBA was found through exome sequencing (Fig. 1). This mutation results in an immediate stop-gain at the amino acid position p.R655* (Fig. 1).
Table 1
Flow cytometry analyses of our patient at 12 years of age
| Lymphocyte subtypes | Cell number mm3 (%) | Normal range mm3 (%) |
|---|---|---|
| CD19 (B cell) | 259 (10) | 219-509 (4.8-24)* |
| CD19+ CD27+ (memory B cell) | 1 (0.5) | 31-152 (9-35)* |
| CD19+ 27+ IGD+ (non-switched B cell) | 3 (2) | 8-81 (3-21)* |
| CD19+ 27+ IGD– (switched B cell) | 2 (1) | 13-72 (4.4-20)* |
| CD19+ 27– IGD+ (naive B cell) | 240 (93) | 128-403 (58-84)* |
| CD3 (T cell) | 1890 (73) | 1200-2600 (60-76)** |
| CD3+ CD4+ (helper T cell) | 190 (10) | 650-1500 (31-47)** |
| CD3+ CD8+ (cytotoxic T cell) | 1380 (73) | 370-1100 (18-35)** |
| CD16+ 56+ (natural killer T cell) | 75 (3) | 100-480 (4-17)** |
The number of hospitalisations and respiratory infections decreased after IVIG therapy. Initially, he received steroid therapy, followed by mycophenolate mofetil. The number of thrombocytopaenia and haemolysis attacks decreased. However, these attacks continued during the follow-up. Therefore, steroid and mycophenolate mofetil therapy were continued. Enteropathy was considered due to recurrent abdominal pain. There were no other gastrointestinal complaints. An abdominal ultrasonography revealed multiple gallstones. A gastroduodenoscopic biopsy revealed chronic atrophic gastritis and partial villous atrophy. Abdominal pain improved after a cholecystectomy (Fig. 2). The patient is now 17 years old, and he is being observed as he undergoes low-dose steroid and IVIG therapies and is in excellent clinical condition.
Discussion
This report presents a patient who was found to have a known mutation in LRBA while being followed up for Evans syndrome and ALPS. We could not obtain clinical information of an LRBA-deficient case with the same mutation as our patient [9]. However, a genotype-phenotype correlation between these patients may not be found in these patients [4, 5] even though they had the same mutation [5]. Multiple gallstones are very rare in children; chronic haemolytic anaemias, inflammatory bowel disease, cystic fibrosis, and biliary tract diseases are predisposing conditions [10]. The presence of AIHA was accepted as a predisposing factor in our patient. Our patient had undergone three AIHA attacks. Two of them had improved by erythrocyte suspension supplementation while the third attack had improved by plasmapheresis. It is easy to correlate the presence of gallstones due to increased haemolysis by AIHA or multiple transfusion. Multiple gallstones have not been reported in other LRBA-deficient patients who also had AIHA and/or IBD [4-9, 11-13]. This report is the first in this respect. There was no other predisposing factor in the patient for multiple gallstones. Therefore, we thought that multiple gallstones in our patient may be an enteropathy finding.
Enteropathy was reported in two-thirds of LRBA-deficient patients in very recent publications [4, 5]. Morbidity seems to be a bit higher in LRBA-deficient patients who manifested with enteropathy during early childhood [11-13]. Alkhairy et al. categorised LRBA deficiency clinically into three phenotypes including autoimmune, immunodeficiency, and enteropathy phenotypes [5]. Enteropathy and lymphoproliferation can be accepted as the findings of immune dysregulation [4]. LRBA-deficient patients usually display more than one phenotype [4-9, 11, 12]. Sometimes only one phenotype or no phenotype may appear in a patient with LRBA mutation [14, 15]. Phenotypes may emerge with increasing age. Our patient developed an autoimmunity, lymphoproliferation, immune deficiency, and enteropathy, respectively. Mild gastrointestinal complaints in our patient emerged during adolescence while taking immunosuppressive agents. As in our patient, partial villous atrophy and atrophic gastritis are reported in LRBA-deficient patients [6, 12]. However, our patient did not have chronic diarrhoea or failure to thrive.
Autoimmunity and immune dysregulation are the most frequent clinical complications in LRBA-deficient patients who have been genetically confirmed [4, 5]. Consistent with the literature, our patient mainly suffered from autoimmune diseases. He suffered for a long time from Evans syndrome, followed by ALPS. The most commonly reported clinical condition accompanying Evans syndrome in children is ALPS, with a rate of 47% [16]. Gamez-Diaz et al. reported that half of patients with definitive LRBA deficiency cases also had ITP and AIHA [4]. Therefore, Evans syndrome can be one of the manifestations of LRBA deficiency [4, 14].
It has been reported that 41-58% of definitive LRBA deficiency patients have immune deficiency, especially CVID [4, 5]. Our patient was diagnosed with CVID at the age of 12, and he had developed autoimmunity at the age of six. We considered possible LRBA deficiency, but we could not test for LRBA mutations at that time. Therefore, the age at diagnosis was late according to the literature [5]. Reduced total B cells, switched memory B cells, plasmoblasts, and Tregs are the most frequent features in flow cytometry analysis [4, 5]. The B cell subtypes of our patient were consistent with the literature. We found an inverted CD4/CD8 T cell ratio, and we inferred this to be a T cell dysregulation.
Conclusions
In conclusion, when immune deficiency and autoimmune syndromes such as Evans syndrome occur together, LRBA deficiency should be considered, especially in cases of ALPS. Gallstones should be considered in the case of intractable abdominal pain in LRBA-deficient patients during adolescence.