
Introduction
Congenital adrenal hyperplasia (CAH) is an umbrella term encompassing seven autosomal recessive genetic disorders pathophysiologically rooted in the dysfunction of the steroidogenesis pathway, resulting in glucocorticoid deficiency [1, 2]. Most commonly it is associated with 21-hydroxylase deficiency (21-OHD), which accounts for more than 90% of CAH cases. CAH may be, in rarer cases, caused by other mutations in genes encoding enzymes in pathways involved in cortisol biosynthesis: 11β-hydroxylase, 3β-hydroxysteroid dehydrogenase type 2 (3β-HSD2), 17α-hydroxylase, P450 oxido-reductase, steroidogenic acute regulatory protein and cholesterol side-chain cleavage enzyme (P450scc) [3, 4]. Depending on the position of the inactivating mutation in enzymes involved in the steroidogenesis, mineralocorticoid and androgen production may be decreased, increased, or unaffected. As a consequence of reduced cortisol levels, the hypothalamic–pituitary–adrenal axis, lacking negative feedback, causes continuous stimulation of the adrenal cortex by adrenocorticotropic hormone (ACTH), resulting in adrenal hyperplasia and enhanced activity of pathways upstream of the enzymatic block [2]. An increased level of ACTH present during the fetal life of 21-OHD patients causes virilization of external genitalia in fetuses with a 46,XX karyotype [5]. Clinically, three types of 21-OHD CAH are distinguished: classic salt-wasting (SW), classic simple virilizing (SV), and a non-classic (NC) form, which is believed to be one of the most common autosomal recessive disorders [6]. SW-CAH occurs when less than 2% of 21-hydroxylase activity is preserved, and can manifest as a life-threatening SW crisis with severe hyponatremia and hyperkalemia, failure to thrive, feeding problems, and shock [6, 7]. The disease may be suspected in female newborns due to the presence of ambiguous genitalia, in both SW and SV forms, prompting further diagnostics. In male patients, however, SW crisis is often the first manifestation, which contributed to higher mortality in the pre-screening era [7, 8]. SV-CAH occurs when slightly higher residual enzyme activity is present, and it does not cause ionic imbalances leading to SW crises [6]. In NC-CAH, up to 50% of normal 21-hydroxylase activity is preserved, causing less obvious symptoms, and therefore the diagnosis is often made in adulthood or omitted completely.
CAH is one of the most demanding endocrine disorders in terms of diagnosis, management, and treatment, due to its rarity and rapid course. It presents a significant challenge to capture, before severe symptoms occur. The introduction of newborn screening program (NSP) allowed us to meet those needs. The neonatal screening for classic CAH due to 21-OHD was first introduced in Alaska in 1977, followed by more than 40 countries worldwide, including all other US states [4, 9]. Poland first introduced newborn screening for CAH in selected voivodeships in 2015, and since the beginning of 2017 it has been applied throughout the country [10, 11, 12]. The reported CAH occurrence frequency in the first years of neonatal screening in Poland is estimated at 1:14,781 [11, 12], while the incidence determined by neonatal screening in most populations is approximately 1:14,000–1:20,000 [1, 7, 13]. Moreover, Suwanlikit et al. [14] described an estimated CYP21A2 gene pathogenic variant carrier frequency of 1 in 65 in the tested population. Despite conducting NSP based on 17-hydroxyprogesterone (17-OHP) immunoassays on dried blood spots, some of the scarcer CAH types remain undetected until the expressed symptoms lead to diagnosis later in life [15].
Screening programs in most countries are focused on detecting classic SW and SV-CAH and preventing severe complications and deaths [8]. However, a wider scope of screening has been already suggested, with potential implementation of genetic testing of populations, enabling proactive prevention of CAH at the preconception stage by identifying carriers who could be provided with genetic counseling and selecting embryos via preimplantation genetic techniques to prevent affected births [14]. However, further research and development of these methods to improve their sensitivity and accuracy, as well as securing funding, is necessary for widespread application.
This study focuses on analyzing the effects and identifying challenges in the first few years after implementing the NSP for CAH in the Polish population.
Material and methods
The study was conducted as a retrospective evaluation of newly diagnosed patients with classic SW-CAH, identified based on biochemical, clinical, and routinely assessed genetic criteria. It included all 23 patients diagnosed in the Małopolska region since the start of the NSP in Poland in 2017. For the statistical analysis, a random equally sized group of 23 patients was chosen from the pre-screening (PRE-SC) population treated in our hospital before 2016. Overall, the study examines patients diagnosed from 1.01.1994 until 1.03.2025.
The inclusion criteria were ICD-10 code E25.0 and positive testing for CAH in the urinary steroid profile. Patients with NC-CAH were excluded from the study. We also collected anthropometric data, as well as the following: age at hospital admission and diagnosis, assigned sex at birth, ion blood levels, urinary and blood steroid profile, evidence of SW symptoms, and hormonal parameters.
All newborns in Poland are screened for 30 congenital metabolic and genetic disorders as a part of the state health policy program commissioned by the Ministry of Health. The NSP is conducted free of charge and is coordinated by the Institute of Mother and Child in Warsaw [12]. The procedure starts by collecting dried blood spots on filter paper, usually on the third day of life. Dried blood spots are then evaluated for 17-OHP by fluoroimmunoassay (FIA), taking into account the gestational age and day of sampling. When an elevated 17-OHP concentration is detected, it is verified by blood steroid profile using liquid chromatography–tandem mass spectrometry at the Institute of Mother and Child in Warsaw. The steroid profile assesses 17-OHP, cortisol, 21-deoxycortisol (21-DF), 11-deoxycortisol and androstenedione concentrations, as well as ratios of (17-OHP + androstenedione)/cortisol and (17-OHP + 21-DF)/cortisol. In the case of preterm newborns, a retest of the second filter paper is required. Afterwards, children with positive screening results are admitted to an endocrine ward and undergo further genetic analysis and treatment [11].
Twenty-four-hour analysis of steroid profile in urine was performed in the Department of Clinical Biochemistry of the Children’s Memorial Health Institute in Warsaw. The analysis was performed using a Hewlett-Packard HP 6890 Series GC System gas chromatograph equipped with a Hewlett-Packard 5973 Mass Selective Detector and a 12-meter HP Ultra 1 fused silica capillary column (Hewlett-Packard). Peak identification was based on comparison of the retention times of the observed peaks with those of steroid standards. Quantitative calculations were performed by comparing the peak areas of the detected steroid standards with the area of the internal standard peak (stigmasterol). Following gas chromatography–mass spectrometry (GC/MS) analysis of the sample, a chromatogram is obtained, which graphically represents the detector signal intensity as a function of retention time. The resulting mass spectrum is characteristic of a specific chemical compound. The mass spectrometer is synchronized with computer-based data processing software, enabling comparison of the acquired mass spectrum with a reference library of known mass spectral patterns corresponding to compounds with established chemical structures. Using this GC/MS technique – designed for the separation and identification of mixture components – a steroid profile analysis was conducted, encompassing 38 steroid metabolites. In cases where precise separation of compound mixtures proves challenging, the method of selected ion monitoring (SIM) is employed. The SIM technique offers substantially greater sensitivity and selectivity compared to full-scan acquisition, which detects all ions resulting from the fragmentation of a given chemical compound.
Data processing and statistical analysis were performed using Statistica 13 PL software. Statistical significance was set at p < 0.05. Normality was assessed using the Kolmogorov–Smirnov and Lilliefors tests. Variables with a normal distribution were analyzed using t-tests and analysis of variance (ANOVA) and reported as mean ± standard deviation; those without normal distribution were analyzed using the Mann–Whitney U test and Kruskal–Wallis ANOVA and reported as median with interquartile range (IQR). The χ2 test was used to evaluate frequency data.
The study was conducted in accordance with the Declaration of Helsinki and approved by the Jagiellonian University Ethics Committee (approval number 1072.6120.120.2022, received on 14.12.2022). It was not necessary to obtain consent from participants or legal guardians because all the data were obtained retrospectively and are fully anonymized.
Results
The study included 46 patients divided into two even groups: the pre-screening group (PRE-SC: n = 23, 46,XY – 6/23, 26%) and the screening group (SC: n = 23, 46,XY – 14/23, 60%). The populations did not significantly differ in terms of birth length, standard deviation score (SDS) birth length, or body surface area (BSA); however, there was a significant difference in terms of both birth weight and SDS birth weight (Table I). The median birth weight was 3250 g in the PRE-SC group and 3630 g in the SC group (p = 0.0161), while the mean SDS birth weight was –0.532 ±0.647 g and 0.142 ±0.857 g, respectively (p = 0.0045). In the PRE-SC group, all but two patients were carried to term (34 weeks of gestation (wk) and 36 wk), and in the SC group only one patient was born prematurely (30 wk). In terms of sex assignment at birth, all 46,XY patients in both groups were assigned male, while 46,XX patients had undetermined sex in 9/17 cases in the PRE-SC group and 7/9 in the SC group. Virilization was observed in all 46,XX cases in both groups (Table II).
Table I
Population characteristics
Table II
Sex assigned and Prader scale at birth
| PRE-SC | SC | |||
|---|---|---|---|---|
| 46,XX | 46,XY | 46,XX | 46,XY | |
| Sex assigned at birth | ||||
| Male | 1 | 6 | 0 | 14 |
| Female | 7 | 0 | 2 | 0 |
| Undetermined | 9 | 0 | 7 | 0 |
| Prader scale | ||||
| I | — | — | — | |
| II | 1 | — | 2 | — |
| III | 5 | — | 3 | — |
| IV | 9 | — | 4 | — |
| V | 1 | — | — | |
Diagnosis, confirmed by 24-hour steroid profile in urine, in the PRE-SC population took a median of 21 days (IQR: 14.00–45.50) (Table III). When grouped by sex assigned at birth, the undetermined (U) population showed the fastest diagnosis at a median of 14 days (IQR: 13.00–16.00), followed by the female (F), 21.00 days (IQR: 13.50–27.00), and male (M), 52.00 days (IQR: 43.50–55.50) – populations (p = 0.0066). Hydrocortisone (HC) supplementation was initiated at a median age of 14 days (IQR: 9.25–22.25), and fludrocortisone (FC) supplementation started at a mean age of 23.33 ±15.18 days. During the first confirmatory hospitalization, the median maximal HC dose per BSA was 89.11 mg/m2/day (IQR: 70.38–95.47), the median maximal FC dose was 50.00 mcg/day (IQR: 50.00–75.00), and the mean maintenance HC dose per BSA was 66.77 ±20.28 mg/m2/day.
Table III
Diagnosis and treatment in pre-screening population
In the SC population, the screening test was sent at a median age of 2.5 days (IQR: 2–3), and the results were received at a median age of 7 days (IQR: 6–8) (Table IV). In 13 patients discharged without abnormalities from the postnatal ward, the screening test was the primary reason for confirmatory hospitalization. Of the other patients, two received the screening results before discharge and were subsequently transferred to University Children’s Hospital in Krakow (the reference center), and 5 were transferred due to ambiguous genitalia (Figure 1). Confirmation of diagnosis based on the steroid profile occurred at a median age of 8 days (IQR 7.5–9.5). Cases with undetermined (U) sex at birth reached diagnostic confirmation earliest, at a median age of 4 days (IQR 3–5), followed by males (M) at 8.5 days (IQR 8.0–9.25) and two female patients (F) at 66.5 days (IQR 50.25–82.75) (p = 0.0028). In both female patients, subtle virilization was noted; across three consecutive follow-up screens, 17-OHP and 21-DF rose progressively. The diagnosis was ultimately confirmed by genetic testing, which identified 21-hydroxylase deficiency in these patients, accounting for the prolonged diagnostic course. HC supplementation was initiated at a median age of 9 days (IQR: 7.25–11.00), and FC supplementation started at a median age of 9.5 days (IQR: 7.25–11.00). Based on the perceived sex at birth, the aforementioned tendency held for the initiation of treatments; first were the U, then M, and lastly F (Table IV). The median maintenance HC dose per BSA was 34.10 mg/m2/day (IQR: 31.25–45.45), and the median maintenance FC dose was 125.00 mcg/day (IQR: 75.00–150.00).
Figure 1
Reasons for hospitalization in screening (SC) population. Of the 13 patients discharged without abnormality, 12 were male and only 1 was female
CAH – congenital adrenal hyperplasia
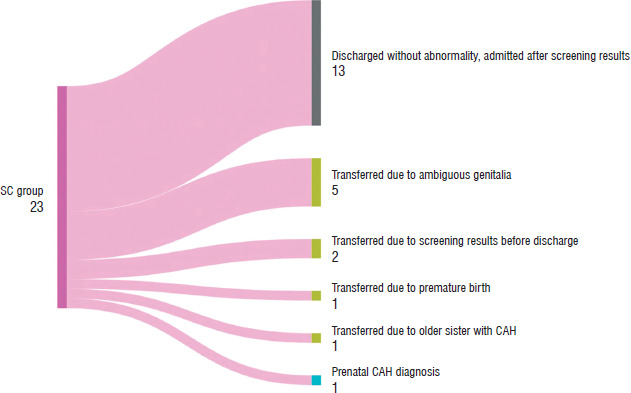
Table IV
Diagnosis and treatment in screening population
Patients in the SC group received the HC and FC treatment earlier than in the PRE-SC group, at a median age of 9 vs. 14 days (p = 0.0175) and 9.5 vs. 19 days (p = 0.0032), respectively. The maintenance HC doses at discharge were significantly lower in the SC group, while the FC maintenance doses at discharge were higher (38.77 ±11.98 vs. 66.77 ±20.28, p < 0.0001 and 125.00 [IQR: 75.00–150.00] vs. 50.00 [IQR: 50.00–75.00], p = 0.0001, respectively) (Table V). The PRE-SC group achieved lower minimal sodium (122.67 ±8.78 vs. 131.43 ±3.89, p = 0.0001) and higher maximal potassium (8.42 ±1.40 vs.7.19 ±1.14, p = 0.0027) concentrations. Additionally, the proportion of patients who suffered from severe hyponatremia (defined as Na concentration < 130 mmol/l) and severe hyperkalemia (defined as K concentration > 7 mmol/l) was significantly higher in the PRE-SC population, at 83% vs. 43% (p = 0.0113) and 91% vs. 62% (p = 0.031), respectively. The SC population achieved their lowest sodium and highest potassium levels earlier (Na: 8 vs. 14.5 days after birth, p = 0.0003, K: 8 vs. 15 days after birth, p = 0.0087), as they were hospitalized earlier; however, the correction to normal levels took them longer than in the PRE-SC group (Na: 11.5 vs. 4 days, p = 0.0008, K: 14 vs. 7 days, p = 0.0207), which can be explained by lower doses of steroids used in this group (Table VI). The dosage of salt replacement therapy was higher in the SC group than in the PRE-SC group (3.24 [IQR: 2.40–3.82] vs. 1.15 [IQR: 0.00–2.66] mmol/kg/day, p = 0.0072). Finally, the duration of hospitalization required to establish the diagnosis of CAH was significantly longer in the PRE-SC group (23.5 days [IQR 15.0–38.0]) than the SC group (15.0 days [IQR 12.5–19.0], p = 0.0197).
Table V
Treatment comparison between pre-screening (PRE-SC) and screening (SC) populations
Table VI
Electrolyte imbalances comparison between pre-screening (PRE-SC) and screening (SC) populations
Discussion
CAH, as one of the most difficult endocrine disorders in diagnostic, management, and treatment aspects due to its rarity and rapid course, presents a challenge to capture it before symptoms occur. When diagnosed too late or left untreated, it may manifest as a life-threatening condition. In SW-CAH, a SW crisis appears most often in the third week of life, ranging from as early as the first days of life to as late as a few months of life, with severe hyponatremia and hyperkalemia, metabolic acidosis, hypoglycemia, dehydration and shock [7, 16]. The first symptoms occur already in an advanced stage of steroid imbalance and involve feeding problems, vomiting, diarrhea, hypotension, a weak cry, weight loss and failure to thrive [16]. If left untreated, it can result in serious complications including central nervous system impairment, circulatory failure, and even death. Another less urgent, albeit serious and important problem, arises in the form of incorrect gender assignment and gender confusion, as well as risk of premature pubarche and short stature in children with SV-CAH [16]. Taken together, these findings highlight that implementing effective strategies to prevent severe, and potentially life-threatening, complications of undiagnosed CAH remains a major challenge. In response, the neonatal screening for classic CAH due to 21-OHD was first introduced in Alaska in 1977, followed by more than 40 countries worldwide, including all other US states [4, 9]. Poland first introduced newborn screening for CAH in selected voivodships in 2015, and since the beginning of 2017 it has been applied throughout the country [10, 11, 12]. Here, we present the first report of neonatal screening for CAH in the Małopolska region.
Diagnosis
Before the advent of the NSP, patients admitted to our center with clinical features suggestive of CAH were first evaluated by measuring serum 17-OHP. The diagnosis was then confirmed by a 24-hour urinary steroid profile, followed by genetic analysis of the CYP21A2 gene. Currently, when neonatal screening results for CAH are positive, the Endocrine Society Guidelines recommend referral to a pediatric endocrinologist and, if the diagnosis remains uncertain, evaluation with a cosyntropin stimulation test [17]. Genetic testing is necessary if the laboratory results are still ambiguous or cosyntropin stimulation cannot be accurately performed [17]. In older children with symptoms suggesting CAH, an early-morning baseline serum 17-OHP measurement by liquid chromatography–tandem mass spectrometry is advised, and only when the result is inconclusive, a steroid profile after a cosyntropin stimulation test is performed to distinguish 21-hydroxylase deficiency from other steroid pathway enzyme defects [17]. In our center, in cases of positive neonatal screening results for CAH, only a 24-hour-urinary steroid profile and genetic testing are performed in order to confirm the diagnosis.
Despite an extensive history of NSP in many countries, sources about its influence on clinical outcomes remain scarce. Some studies from Canada and New Zealand reported a significantly shorter time to diagnosis after the introduction of SC, while others, including studies from Lithuania and the USA, did not observe a significant difference in time of diagnosis [18, 19]. Analyzing these sources, we inferred that a crucial factor influencing the time from birth to diagnosis and introduction of treatment, and thus determining the effectiveness of NSP, is the time between collecting and delivering the samples and receiving the results [18]. In most national neonatal screening programs, blood samples are obtained after 72 hours of life. However, in some countries, such as the United States, samples are collected at 48 hours to allow earlier detection of other serious inborn errors requiring immediate intervention to prevent irreversible complications [20]. The challenge in optimizing the screening timeline lies in balancing the acute nature of other diseases included in NSP and an increased rate of false positive results for CAH screening connected with earlier sampling, as 17-OHP levels tend to be physiologically elevated in the first days of life.
In the present study, we found that the median time from birth to collecting samples in our population was 2.5 days and from birth to obtaining results was 7 days (Table IV). We concluded that the time to diagnosis was significantly shortened by introducing NSP in Poland, as the time from birth to diagnosis in PRE-SC was a median of 21 days, and the time of obtaining confirmation of diagnosis by steroid profile in dried blood in SC was at a median age of 8 days. Before the introduction of screening or NSP, there were differences between sex groups: patients with unassigned sex at birth were diagnosed the fastest (14 days), and boys were not diagnosed until the 52nd day of life.
In our study, we observed that children in the SC group had a greater birth weight than those in the PRE-SC group. However, there were 2 patients born prematurely in PRE-SC and 1 in SC, but in an earlier stage of pregnancy (PRE-SC: moderately preterm and late preterm; SC: very preterm stage). All 46,XX patients in our study were virilized, but there were more girls in PRE-SC (17) vs. SC (9). We found that 7 out of 9 SC patients with 46,XX karyotype had unassigned sex at birth, while it occurred in only 9 out of 17 PRE-SC patients.
There are studies stating that due to low efficiency of NSP in preterm newborns, screening should be omitted in that group [6, 21]. On the other hand, Gidlöf et al. [22]. favor neonatal screening for CAH in premature patients over sole supervision on the neonatal ward, because of the possibility of earlier detection of affected patients and easy access to a second blood sample for confirmation of screening results. Furthermore, evaluation of body mass changes after birth is considered to be another valuable tool for predicting risk of SW and CAH [20].
Diagnostic challenges
Diagnosis of CAH is a specialized multi-step process. Before the implementation of neonatal screening, CAH diagnosis relied on steroid profiling from 24-hour urine collections using GC/MS. In the absence of CYP21A2 activity, CYP11B1 atypically converts 17-OHP to 21-DF. Therefore, diagnostic ratios of urinary metabolites of 21-DF (pregnanetriolone) or 17-OHP (pregnanetriol and 17α-hydroxypregnanolone) relative to glucocorticoid metabolites were considered highly valuable for confirming CYP21A2 deficiency. It is noteworthy that Poland has a unique experience of more than 40 years in analyzing the results of 24-hour urinary steroid profiles.
With regard to plasma or serum steroid metabolite analysis, in our center we are limited to the measurement of 17-OHP, dehydroepiandrosterone sulfate, ACTH, cortisol, and plasma renin activity. However, until 2024, due to financial constraints, dilution of 17-OHP samples could not be performed beyond concentrations of 15.2 ng/ml, and in some cases even 8.9 ng/ml, which markedly limited diagnostic accuracy. In addition, the turnaround time for serum 17-OHP results still exceeds two months.
Fortunately, with the advent of the NSP, all analyses are now reliably performed on dried blood spots: first-tier 17-OHP measurement by FIA, followed by second-tier short steroid profiling in blood using liquid chromatography–mass spectrometry. This approach has significantly accelerated diagnosis.
Furthermore, the NSP has contributed to shorter hospital stays in the screened cohort compared with the pre-screening era. Earlier discharge reduces the risk of hospital-acquired infections for both children and parents, improves overall well-being, and lowers the economic burden of care. In addition, treatment can now be initiated earlier and with lower glucocorticoid doses.
Clinical presentation
Most studies evaluating the influence of introducing screening for CAH have noted major successes, including a reduction or elimination of mortality and improved clinical outcomes in newly diagnosed cases (Table VII). Although scarce, some sources have estimated that the mortality in the pre-screening era was around 10%, with most deaths related to inadequate administration of stress doses of HC [20, 23, 24]. On the other hand, Hird et al. [25] reported no increase in mortality in the absence of screening for CAH. Children identified through NSP tend to have less severe hyponatremia and shorter hospitalization time [1]. In our study, we observed an improvement in the clinical condition of newly diagnosed CAH patients. The PRE-SC population had lower minimal sodium concentrations (122.67 ±8.78 vs. 131.43 ±3.89) and higher maximal potassium concentrations (8.42 ±1.40 vs. 7.19 ±1.14) than the SC population. The SC group also achieved their lowest sodium and highest potassium levels earlier, although correction to normal levels took them longer than in the PRE-SC group (Table VI). This might be a result of an earlier diagnosis, catching the condition in a less severe state, which therefore required milder forms of treatment. Additionally, in the past, both introductory and maintenance HC doses were higher, whereas currently lower HC doses are preferred, balancing electrolyte disturbances with metabolic complications. Even in the era of screening, physicians should still be alert to potentially life-threating states in newborns with CAH diagnosed through the NSP. In a study conducted in Japan, it was concluded that despite eliminating lethal cases, there are still many screened children (almost 40%) who suffer from severe SW [26]. Therefore, it is important that the necessary interventions should be undertaken as soon as possible, preferably in the first week of life, as in most cases severe hyponatremia or hyperkalemia occurs in the middle of the second week of life (11.1 and 12.3 days, respectively) [20, 27].
Table VII
Literature review of selected studies on the topic of newborn screening program for congenital adrenal hyperplasia
| Country (first author, year of publication) [reference] | Time of collecting samples (days of life) | Time of diagnosis with screening (days of life) | Time of diagnosis before screening (days of life) | Treatment initiation with screening (days of life) | Treatment initiation before screening (days of life) | Positive screening results (days of life) | Number of cases diagnosed in NSP | Mortality with Screening | Mortality before Screening | Abnormal virilization with screening | Abnormal virilization without screening | Gender misidentification with screening | Gender misidentification without screening | Serum sodium at diagnosis with screening, [mmol/l] | Serum sodium at diagnosis without screening, [mmol/l] | Serum potassium at diagnosis with screening, [mmol/l] | Serum potassium at diagnosis without screening, [mmol/l] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| New Zealand (Cutfield 1995) [28] | 4.3 ±1.0 | 11 ±3 | ND | 13 (4-35) | ND | ND | 14 | ND | ND | 13/14 | ND | 1/3 | ND | 124 ±7.2 | ND | 7.8 ±0.7 | ND |
| New Zealand (Heather 2015) [29] | 3.3 (2-8) | 8.5 ±3.2 (3-16) | 11 ±3.0 | 12.0 (6-30) | 14 (0-26) | 5.2 (1–12) | 21 | ND | ND | 7/21 (33%) | 23/23 (100%) | 1/21 (5%) | 0 | 130.8 ±5.2 | 136.8 ±4.1 | 6.02 ±0.78 | 5.29 ±0.95 |
| Canada (Fox 2020) [19] | ND | 6 (0-13) | 5 (0-30) | ND | ND | 5.5 (0-13) | 17 | ND | ND | 2 | ND | 1 | ND | 136 (125-144); 4/17 Na < 132 | 136 (106-145); 17/40 Na < 132 | 6.2 (4.3-8.5) | 6.9 (3.7-10.3) |
| Japan (Tsuji- Hosokawa 2021) [20] | 4-7 | 7,5 | 55 | ND | ND | 6 | None | None | 10.6% | ND | ND | None | 12.9% 46,XX (firstly assigned as males) | ND | ND | ND | ND |
| Sweden (é 1998) [30] | ND | ND | ND | ND | ND | ND | 31 | ND | ND | ND | ND | 1/31 | 6/35 | ND | ND | ND | ND |
| Sweden (Gidlöf 2014) [22] | 4.0 (2.4 earlier sampling procedure) | 8.7 (3.0) | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| USA/Texas (Brosnan 1998) [31] | ND | 9 (1–34), summarized | 9 (1-34), summarized | ND | ND | 13 (9-27) | 8 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| Brazil (Kopacek 2019) [32] | 5 (2.0-38.0) | ND | ND | ND | ND | 19 (8.0-51.0) | 15 | ND | ND | 11/15 | ND | 2/15 | ND | 125 ±8.30 | ND | 6.10 ±1.01 | ND |
| Brazil (de Miranda 2021) [24] | 5.25 (95% Cl, 4.87-5.82) | 17.3 | 38.8 (95% Cl, 31.1-46.4) | ND | ND | 17 | 25 | ND | 11% | ND | 28 (44%) | ND | 10/67 (15%) 46,XX (assigned as males) | 133.7 mEq/l (95% Cl, 131.5-135.8) | 121.2 mEq/l (118.3-124.1) | ND | ND |
| Lithuania (Navardaus-kaitė 2021) [18] | Median 3 | Mean 14.6 ±9.6; median 13.25 (1-32) | Mean 16.5 ±11.6; median 15.7 (0–42) | ND | ND | 13 (7-23) | 12 | ND | ND | ND | ND | 2/12 (2/2 females) | ND | 127.5 ±7.9 | 127.6 ±11.6 | 6.7 ±1.3 | 6.9 ±1.6 |
| Taiwan (Liu 2018) [33] | ND | 11 (6-127) | ND | ND | ND | ND | 26 | ND | ND | 12/12 girls | ND | ND | ND | 131 mmol/l (119-135 mmol/l) | ND | 6.1 mmol/l (4.5-8.2 mmol/l) | |
| Spain (Sanz Fernández 2023) [34] | ND | 8.5 (6-10.5) | ND | 8 (6-9) | ND | 46 | ND | ND | 12/14 girls | ND | 6/14 girls | ND | 128.3±7.5 | ND | 6.7 ±1.3 | ND | |
| Germany/Bavaria (Odenwald 2015) [35] | 2 (0-5) | 5 (1-10) | ND | 7 (1-112) | ND | ND | 60 | ND | ND | 3 girls | ND | ND | ND | ND | ND | ND | ND |
| Sri Lanka (Seneviratne 2021) [36] | ND | ND | 20 | 30 boys,10 girls | ND | ND | ND | ND | 96% girls | ND | ND | ND | ND | ND | ND | ||
| Turkey (Güran 2020) [37] | ND | ND | ND | ND | ND | 17.35 ±5.64 (7-54) | 20 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| France (Coulm 2003) [21] | Median 4 | 7(1-10) | ND | ND | ND | ND | 358 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
Treatment
Generally, lifelong replacement doses of glucocorticoids are the main method of treatment in CAH, with HC being recommended for children during the growing period [17]. Newly diagnosed infants should have a treatment regimen adjusted to the clinical state. In cases of relevant ionic imbalances, the therapy should be implemented immediately, and greater doses are needed. According to the regimen proposed by Lajic et al. [5], the initial HC dose should consist of an intravenous 5 mg/kg HC bolus, followed by 25 mg per day divided into 3–4 doses and accompanied by intravenous glucose and sodium infusion, if needed. If the child is stable and without electrolyte imbalances, treatment may consist of just oral HC, administered three times a day in 5 mg doses, and after few days a decrease to 2.5 mg and 1 mg doses should be considered. In SW-CAH, due to mineralocorticoid deficiency, FC is also a key element of therapy. In the newborn and infancy period, additional introduction of salt supplementation with 10% sodium chloride is advised [5, 17]. When the final growth is achieved, patients may be maintained on hydrocortisone, or if necessary converted to glucocorticoids of greater strength or their long-acting forms if available [17].
Another improved aspect of CAH care after introducing NSP is the shorter time from birth to the start of treatment. We observed that in the PRE-SC population HC and FC supplementation was initiated at a median of 14 days and a mean of 23.3 days of life, respectively, while in SC children it was a median age of 9 days to start HC and 9.5 days to start FC. Moreover, the mean maintenance dose of HC decreased from 66.77 mg/m2/d in the PRE-SC group to 34 mg/m2/d in the SC group. Interestingly, the maximum HC dose in the PRE-SC group reached as high as 89.1 mg/m2/d, which is markedly higher than the previously advised doses for newly diagnosed newborns.
NSP outlooks
Despite the almost 50 years of CAH screening program history, some issues remain uncertain. One of them is the timing of specimen collection, pointed out by Heather and Nordenstrom, as a balance has to be struck between early detection, prioritizing the diagnosis of the most severe cases before SW symptoms appear and later testing, which could possibly identify more cases, including less severe ones [8]. Another potential concern according to the authors is the relatively high false positive rate and low positive predictive value of CAH screening. This is mostly due to the broad variation in 17-OHP levels in the neonatal period and in severely ill infants, as well as falsely elevated levels in preterm infants, as a result of cross-reactivity with metabolites found in the immature adrenal glands [8]. Additionally, they noted that the potential lack of clinical follow-up and limited availability of treatment in some countries pose further challenges for neonatal screening worldwide [8]. However, it is believed that introducing screening programs in many countries contributes to lowering, or even eliminating, mortality among CAH patients in neonatal age [5].
Another interesting group, which requires closer attention in terms of early detection, comprises patients with rarer forms of CAH. Despite their exclusion from the current study, we observed that among the 3 patients with CAH due to 3β-HSD2 deficiency who remain under our care, the one diagnosed through the NSP has been in a better clinical condition [38]. However, in our experience, not all rare forms of CAH are caught in the NSP; for example, cholesterol P450scc deficiency and aldosterone synthase deficiency evade the detection process due to normal levels of 17-OHP [15, 39]. In both cases, patients were diagnosed in critical condition due to adrenal crises and SW crises in cholesterol P450scc deficiency and SW crises in aldosterone synthase deficiency. In order to cater to patients with rarer forms of CAH, the NSP could strive to broaden the spectrum of analyzed steroid metabolites.
Some have suggested extending the preventive interventions into the prenatal period by screening for pathogenic variants in the CYP21A2 gene, with various laboratories offering preconception panels, giving possibilities for prenatal diagnosis and embryo selection via preimplantation genetic techniques to prevent affected births [14]. The European Molecular Genetics Quality Network recommends applying at least two molecular genetic testing methods for CAH patients, involving complete gene DNA sequencing and multiplex ligation-dependent probe amplification for copy number detection [14]. For the purposes of screening, more rapid and cost-effective methods such as qPCR and PCR-based restriction fragment length polymorphism proved appropriate [14]. However, further research and development of these methods to improve their sensitivity and accuracy is necessary for widespread application.
Long-term consequences
Some studies mentioned a possible influence of clinical presentation in the neonatal period, especially severity and duration of hyponatremia, on future neurological development and intellectual functioning [18, 27]. Imbalances in hormonal levels, repeated episodes of hypoglycemia, and salt-losing crises may also affect brain structures and cognitive abilities [5]. Moreover, avoiding treatment with extremely high stress doses of glucocorticoids in the neonatal period may be an important factor [5]. Taking all the above into account, screening is an essential point in the care of CAH patients, preventing potential negative effects on the brain and development.
Strengths and limitations
We consider the principal strength of our study to be the first-ever comprehensive analysis of the clinical characteristics of all detected cases of CAH in the Małopolska region since the introduction of newborn screening in Poland. This analysis demonstrated a substantial positive impact of screening on the management of our patients, which is likely to translate into improved long-term outcomes, such as greater final height and a reduced risk of metabolic complications.
The main limitations of this study include the relatively small sample size in both cohorts, which reduced the statistical power of the analyses, the random allocation of patients to the PRE-SC group, and the retrospective design.
Conclusions
Despite a relatively short history of NSP or screening for CAH in Poland, we have already achieved an improvement in the level of care in this patient population. Children diagnosed through the NSP present in better overall clinical condition, with less severe electrolyte imbalances, require lower doses of glucocorticoids, and are better metabolically stabilized. Their parents receive education earlier and experience less stress, as the children’s general condition is better than it was before the implementation of the NSP. Nevertheless, there is a lot of work to do: screening programs require constant evaluation and quality improvement, and the diagnostic process in the future would ideally start in the preconception or prenatal period. A valuable aspect of the care process would be improved access to better treatment options, e.g. HC in the form of granules in capsules (Alkindi), providing flexibility to clinicians in tailoring the treatment, and a national CAH registry assessing long-term outcomes for patients diagnosed through screening. Moreover, improved access to educational materials on salt wasting and adrenal insufficiency is essential to prevent serious complications, particularly among parents, healthcare professionals, and primary care institutions.









