

Introduction
Higher rates of mortality, impairment and morbidity are associated with cerebral ischemia, which contributes to almost 80% of stroke events, which makes cerebrovascular disorders a significant contributor among three devastating diseases [1]. Adaption of modern lifestyles by adults and the elderly have exhibited an increase in the incidence of hypertension, elevated blood cholesterol, cardiovascular complications, diabetes, atherosclerosis and diseases associated with the cerebrovascular system [2]. Cerebral infarction results in discontinuance of blood to the brain; thereby glucose and oxygen supply to the ischemic site is impeded. Urgent supply of blood to the ischemic area will significantly lower the ensuing neuronal damage [3]. However, an investigation has revealed that reperfusion may progress to cerebral ischemia-reperfusion injury [4]. A moderate episode of cerebral ischemia hampers learning and memory functionality, which leads to vascular dementia and further makes the individual susceptible to Alzheimer’s disease [5].
During events of oxidative stress, the mitochondrial membrane potential weakens, which leads to splitting of mitochondria. The surface of mitochondria bears Bax and Bak dimer pores, which are known to promote the release of cytochrome C and potentiate the caspase proteins that modulate the process of apoptosis in mitochondria [6]. The mitochondrial fission is modulated by two crucial proteins, namely dynamin-related protein-1 (Drp1) and fission-1 protein (Fis1) [7]. Both cytoplasm and mitochondria contain Drp1 and include the GTPase receptor and GTPase effector, whereas the Fis1 protein resides in the outer region of the mitochondrial membrane. Drp1 is recruited from nearby mitochondrial and cytoplasmic areas at the fission initiation site. Under normal condition, mitochondrial fission does not occur when Drp1 is present in the cytoplasm in a dormant form. The activated form of Drp2 splits the mitochondrion by formation of a helical tubular structure at the axial end of the parent mitochondrion, which leads to mitophagy. Hence, the activation of Drp1 is a crucial step for cleavage of mitochondria [8]. Mitophagy removes the broken mitochondria and restricts these organelles from triggering the mitochondrial apoptotic process [9].
Together LC3 and sequestosome 1 (SQSTM1) are regarded as important markers in the identification of damaged neurons at the ischemic site [10]. The extent of autophagy is marked by the presence of LC3 on the target membrane, and hence it is used as an essential marker. Beclin-1 positively modulates the process of autophagy. A drop in mitochondrial membrane potential stimulates the mitophagy. Bcl2 interacting protein-3 (Bnip3) attracts LC3 on the damaged mitochondrial membrane, thereby inducing mitophagy. PTEN-induced putative kinase 1 (PINK1) is a serine/threonine kinase and is interlinked with inner and outer membrane regions [11].
Several models of ischemic cerebrovascular disease are used, including bilateral common carotid artery occlusion (BCCAO), cerebral ischemia, middle cerebral artery occlusion ischemia/reperfusion and ischemic-hypoxic brain injury [12]. In the BCCAO model, the cerebral blood supply is impeded by 50-60%, which leads to considerably reduced cerebral perfusion. Hippocampal and the cortical regions are areas prominently affected by such hypoperfusion, which significantly influences learning and memory efficiency, and hence such a model is favorable for mimicking vascular dementia.
Pharmacological activities mediated by xanthones include anti-inflammatory, anti-cancer, anti-thrombotic and anti-microbial properties [13]. Euxanthone (EUX), a xanthone derivative isolated from the plant Polygala caudata, has been reported to promote neurite outgrowth [14]. EUX remarkably attenuates memory and spatial learning impairment caused by Aβ1-42 and apoptosis and reverts the neuroapoptosis in hippocampal and cortical regions. EUX also reduces oxidative stress and ROS generation caused by Aβ1-42 in a rat model [15]. As there were no reports earlier, we investigated the therapeutic effect of EUX in cerebral ischemia and reperfusion injury by employing a BCCAO animal model.
Material and methods
Animals
Forty male ICR mice (20 ±2 g) were procured from the First Affiliated Hospital of Wannan Medical College, Anhui, China, and housed in a clean and pathogen-free environment at 25 ±1°C and 65% relative humidity. The animals were allowed free access to food and water and exposed to a 12-hour light and dark cycle. The experimental procedures conducted were recommended and approved by the Research Review and Ethics Board of the First Affiliated Hospital of Wannan Medical College, Anhui, China and the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
Induction of cerebral ischemia and reperfusion
The BCCAO method was conducted for 60 minutes to induce cerebral ischemia and reperfusion in mice. The animals were randomized and divided into four groups – Sham (surgically operated without occlusion using ligated surgical silk), BCCAO (surgically operated and occluded using ligated surgical silk), EUX30 (surgically operated, occluded using ligated surgical silk and treated with 30 mg/kg EUX) and EUX60 (surgically operated, occluded using ligated surgical silk and treated with 60 mg/kg EUX). Both EUX30 and EUX60 group animals received EUX intragastrically dissolved in distilled water once a day for 7 days prior to the surgery until being sacrificed. On the day of surgical intervention, the animals received EUX 2 hours before the cerebral ischemia. The Sham and BCCAO group received distilled water only. The animals were anesthetized in a laboratory anesthesia system (Matrix VIP 3000; Midmark, USA) by initial administration of 4% isoflurane by inhalation, followed by exposure to 1.5% isoflurane in a 7 : 3 nitrous oxide/oxygen mixture. The bilateral common carotid arteries were exposed and then separated followed by occlusion twice for 60 minutes using ligated surgical silk. Between two episodes of ischemia, reperfusion was done for 10 minutes. During the surgery, the animal body temperature was maintained at 37 ±0.5°C using a heater until the effect of anesthesia declined.
Assessment of neurological grading
The neurological deficit grading was recorded, as reported earlier [16], on a 0-5 scale: 0 point – mice with no neurological damage or symptoms, 1 point – minor injury in which the mice was unable to fully extend the contralateral forepaw, 2 points – moderate injury in which the mice circled on the contralateral side, 3 points – serious injury in which the mice fell on its contralateral side, 4 points – the mice did not walk spontaneously or lost consciousness, and 5 points – where the mice did not initiate prompt movement or walking.
Morris water maze test
To determine the space relative learning and memorizing capability of animals, the Morris water maze test was employed. The methodology involved the use of a swimming pool with 100 cm diameter and divided into four quadrants. During the training session, the subject mouse was left within one of the quadrants and was allowed to locate a randomly placed floating platform, placed slightly below the surface of the water. The animal was allowed to rest over the platform for 15 seconds after discovering the floating platform. In case of inability to find the platform within 120 seconds, the animal was manually escorted to the platform and permitted to rest over it for 15 seconds. The subject animals were trained twice daily for five successive days and the escape latency was recorded. The floating platform was removed after the five-day training session, and the time spent by the animal in the target quadrant where the floating platform was based, the distance covered by swimming and the number of times the platform was crossed were registered.
Estimation of hippocampal oxidative markers
The hippocampus was carefully removed and homogenized in normal saline (1 : 10 w/v), and subjected to centrifugation at 2500-3000 rpm in cold for 10 minutes. The supernatants collected were analyzed for protein content by employing a BCA kit (Wuhan Fine Biotech Ltd., China). ELISA (Shanghai Korain Biotech, China) was used for estimation of hippocampal reactive oxygen species (ROS) levels, as per the manufacturer’s instructions. The activity of superoxide dismutase (SOD) and malondialdehyde (MDA) content was also analyzed according to the manufacturer’s instructions (Shanghai Korain Biotech, China). The optical density was determined at 450 nm by employing a microplate reader (Thermo Fisher, IL, USA).
Western blotting
The hippocampus removed from mice was homogenized in cold lysis buffer blended with a protease inhibitor. The homogenate was subjected to centrifugation in cold for 20 minutes at 12,000 rpm. Using a BCA protein assay kit (Wuhan Fine Biotech Ltd., China), the protein concentration was determined in the supernatant, as per the manufacturer’s instructions. These aliquots were fractionated by employing the sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) technique. The separated proteins were loaded on polyvinylidene fluoride membranes (PVDF), which were then blocked with 5% skimmed milk added into tris-buffered saline containing Tween 20 for 2 hours. The PVDF membranes were then treated with primary antibodies (Cell Signalling, Shanghai, China) – Beclin-1 (#4122) (1 : 1000), LC3 (#2775) (1 : 1000), p53 (#2425) (1 : 1000), Bax (#2772) (1 : 1000), caspase-3 (#9668) (1 : 1000), BNIP3 (#3769) (1 : 500), DRP1 (#14647) (1 : 1000), DRP1 Ser616 (#3455) (1 : 500), DRP1 Ser637 (#4867) (1 : 500), β-actin (#3700) (1 : 1000) and incubated for 24 hours at 4°C. The Nrf2 (#12721) (1 : 1000) levels were determined in nuclear and cytoplasmic fractions against Lamin-B (#13435) and β-actin (#3700) (1 : 1000) respectively. The membranes were rinsed three times with tris-buffered saline containing Tween 20 and allowed to rest with secondary antibodies linked with horseradish peroxidase for a further 2 hours at room temperature. The protein fractions were then treated with chemiluminescence reagents and observed using a gel imaging system (Bio-Rad). The bands were quantified in an optical density reader using the ImagePro Plus software package.
Statistical analysis
The data are represented as mean ± standard error of the mean (SEM). One-way ANOVA and Tukey’s post hoc test were performed to verify statistical differences among the groups. For comparisons values of p < 0.05 were considered statistically significant. The statistical analyses were performed using GraphPad Prism software 8 (GraphPad, USA).
Results
EUX ameliorated cognitive impairment in cerebral ischemia mouse model
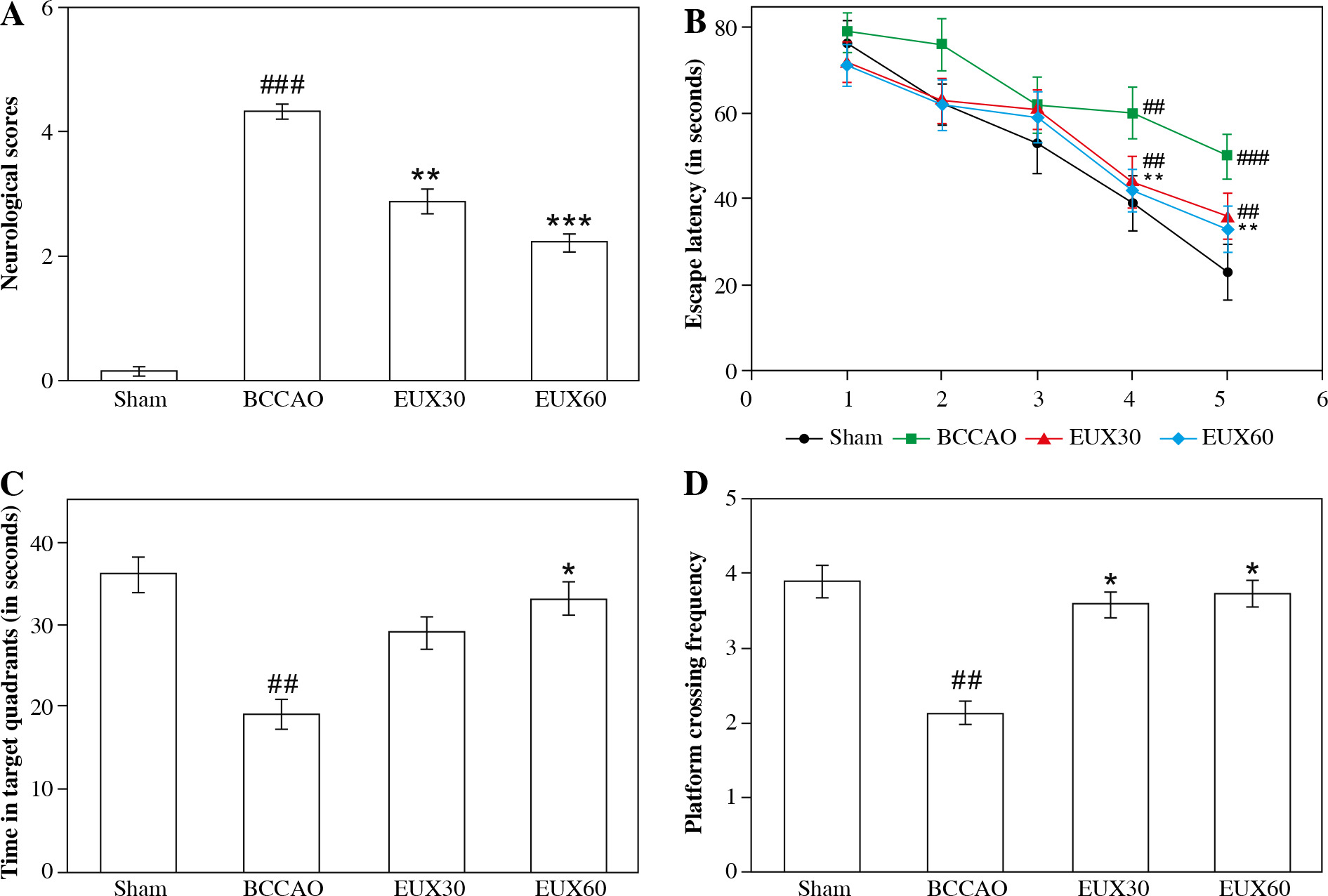
Neurological grade and the Morris water maze test were used to elucidate the effects of EUX on learning and remembrance capacities of BCCAO induced mice. The results (Fig. 1A) revealed significantly poor neurological grade in mice induced with BCCAO, compared to the Sham group. However, the mice treated with EUX (30 and 60 mg/kg) revealed marked improvement in their neurological score, compared to the animals that did not receive EUX. The Sham group animals indicated significantly higher escape latency in the Morris water maze test on the fourth and fifth days of training, compared to the BCCAO group mice (p < 0.01, p < 0.001), which revealed reduced escape latency. These observations demonstrate that learning and memory impairment results after cerebral ischemia and reperfusion in mice. The EUX60 group exhibited significant reduction in the escape latencies compared to the BCCAO group from the fourth and fifth days (p < 0.01) of the training (Fig. 1B). The time spent in the designated zone and the frequency of crossing the platform were reduced in the mice from the BCCAO group (p < 0.01). After treatment with EUX, there was a significant rise in the time spent in the designated quadrant (p < 0.05) and the frequency of crossing the platform (p < 0.05) by the subject mice, compared to the BCCAO group animals (Fig. 1C, D). The results suggest the potential ability of EUX to reduce the cognitive disability caused by short-term cerebral ischemia and reperfusion.
Fig. 1
Effect of EUX pre-treatment on learning and remembrance capacities of BCCAO induced mice was evaluated by estimating the neurological grade and conducting a Morris water maze test. Values are expressed as means ± S.E.M. (n = 8). Compared to the Sham group: #p < 0.05, ##p < 0.01, ###p < 0.001. Compared to the BCCAO group: *p < 0.05, **p < 0.01, ***p < 0.001
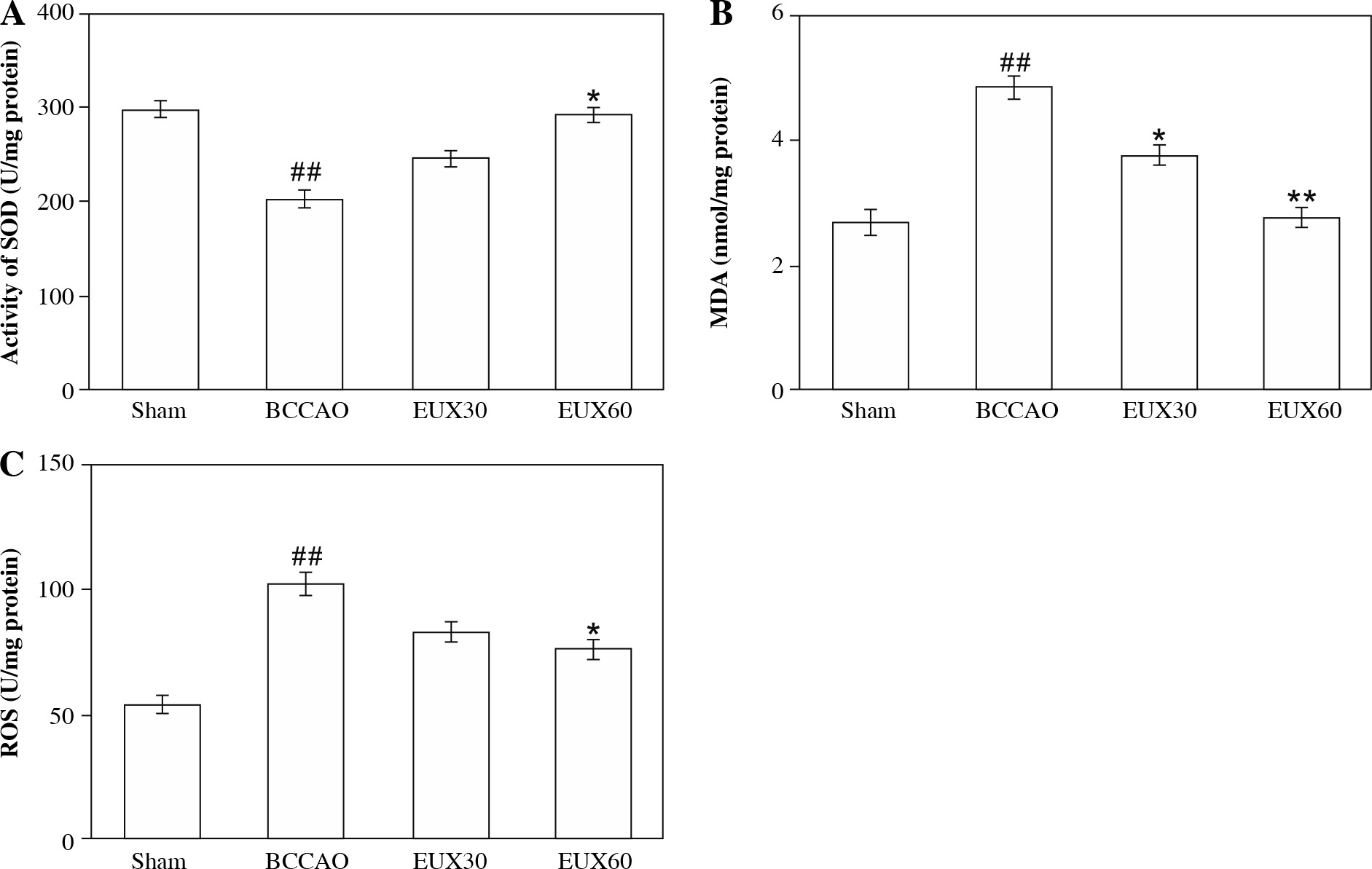
EUX attenuated the hippocampal oxidative stress in BCCAO treated mice
Reactive oxygen species, MDA and SOD are significant markers to estimate the extent of oxidative stress. In the present investigation, hippocampus collected from BCCAO group mice indicated a significantly (p < 0.01) higher MDA level, compared to the Sham group mice. Mice from groups treated with EUX indicated significant (p < 0.01) normalization of the MDA levels, compared to the BCCAO group mice, in a dose-dependent manner (Fig. 2A). SOD level was markedly (p < 0.01) reduced in the animals from the BCCAO group, compared to the Sham mice, whereas mice from groups treated with EUX revealed a significant (p < 0.05) increase in the SOD level (Fig. 2B). Compared to the mice from the Sham group, the level of ROS was significantly (p < 0.01) higher in the BCCAO group. Animals that received EUX indicated a marked (p < 0.05) reversal of the ROS level to normal after induction of cerebral ischemia (Fig. 2C).
Fig. 2
Effect of EUX pre-treatment on the levels of MDA, SOD and ROS was estimated to evaluate the extent of ox- idative stress in the hippocampal fractions. Data represent means ± S.E.M. (n = 8). Compared to the Sham group: #p < 0.05, ##p < 0.01, ###p < 0.001. Compared to the BCCAO group: *p < 0.05, **p < 0.01, ***p < 0.001
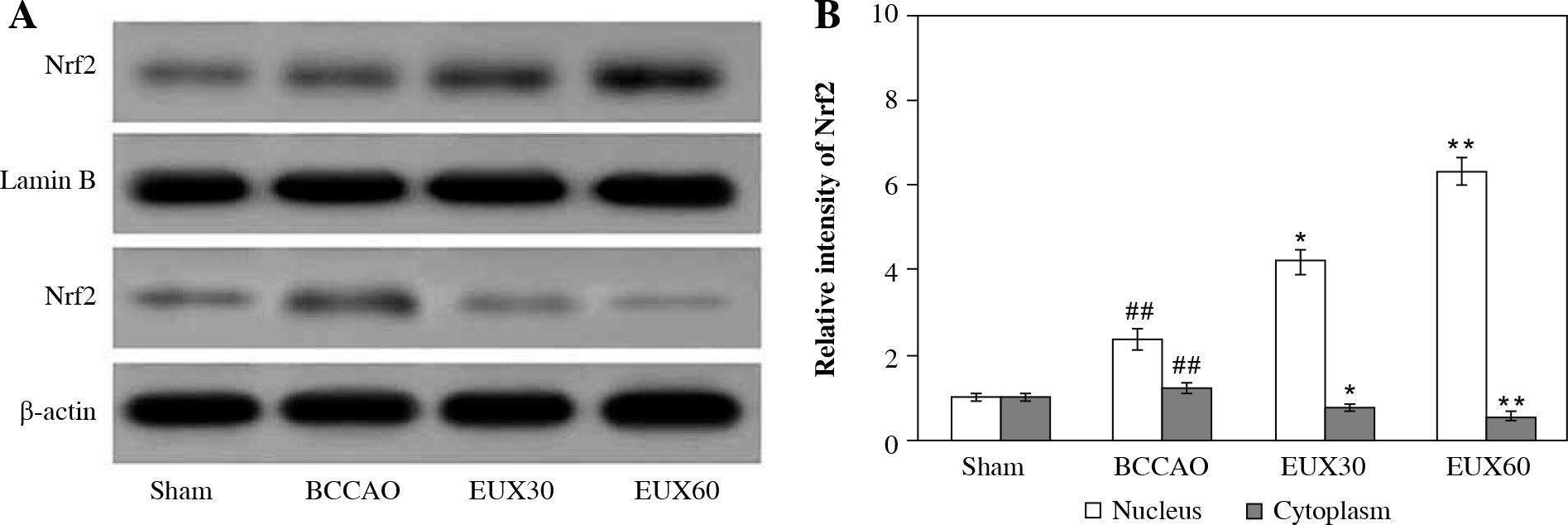
EUX enhanced Nrf2 expression in the nuclear fraction in the hippocampi
The hippocampal sections derived from mice were subjected to immunochemical estimation of Nrf2 expression, and visualized using Western blotting (Fig. 3A). The expression of Nrf2 was significantly (p < 0.01) reduced in the hippocampal sections collected from the BCCAO group, indicating oxidative stress generation due to BCCAO in the hippocampus. EUX (30 and 60 mg/kg) administration significantly elevated the Nrf2 expression in the nuclear fraction and concurrently reduced it in the cytosolic fraction, which indicates that EUX can potentially reduce the oxidative stress caused by cerebral ischemia in mice (Fig. 3B). The results of Nrf2 expression in nuclear and cytosolic fractions from EUX30 and EUX60 groups were statistically significant, with p < 0.01 relative to the Sham group, and p < 0.01 relative to the BCCAO group.
Fig. 3
Effect of EUX pre-treatment on the Nrf2 expression in the hippocampal cytoplasm and nuclear fractions of the hippocampus of BCCAO-treated mice measured by Western blot. Data represent means ± S.E.M. (n = 8). Compared to the Sham group: #p < 0.05, ##p < 0.01, ###p < 0.001. Compared to the BCCAO group: *p < 0.05, **p < 0.01, ***p < 0.001
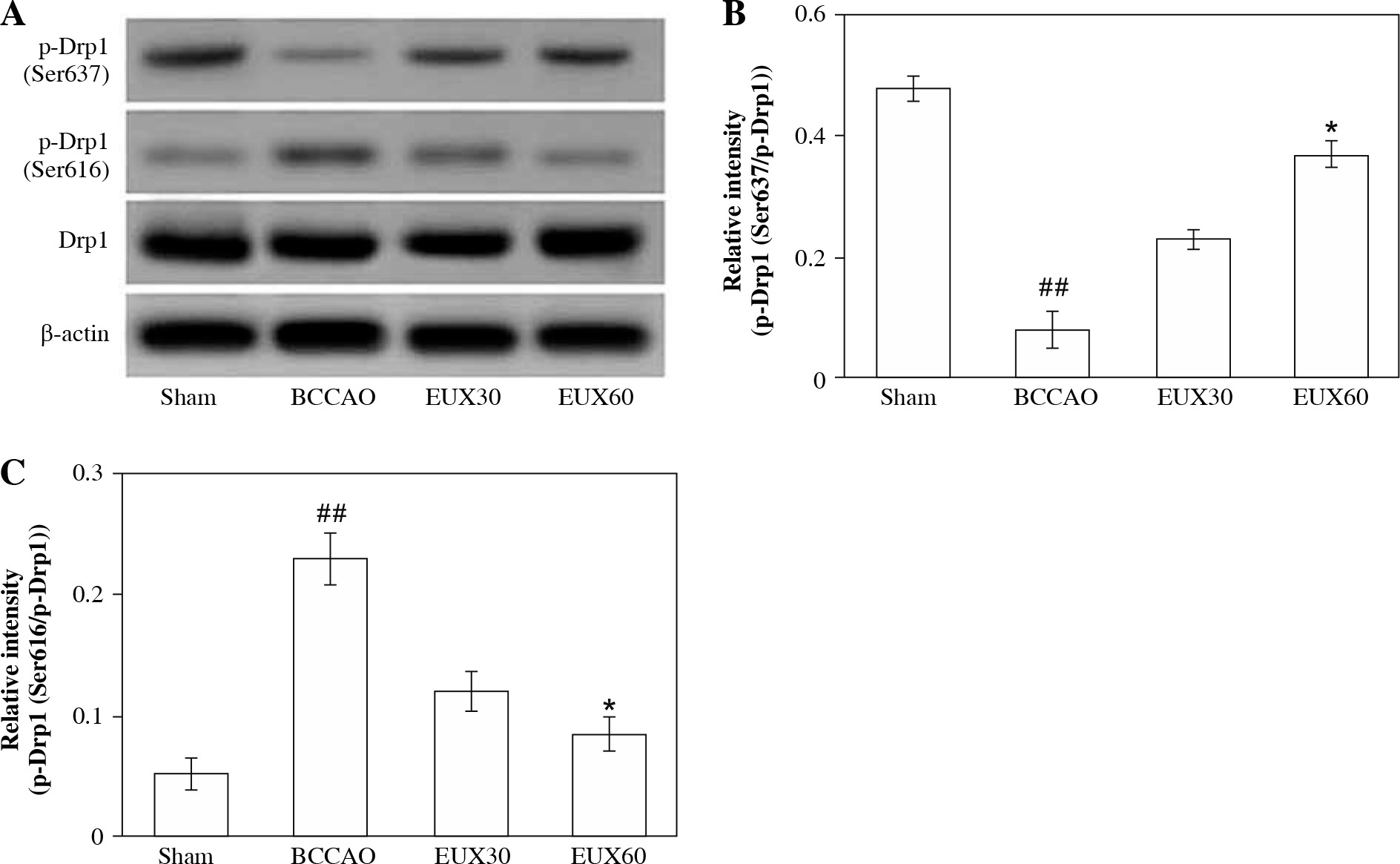
EUX suppressed hippocampal mitochondrial fragmentation in BCCAO treated mice
The results of the Western blot analysis revealed that the hippocampal Drp1 protein content in the mice from the BCCAO group was not markedly altered. However, activated Drp1 (Ser637), that is p-Drp1 (Ser637) expression was significantly (p < 0.01) restrained and activated Drp1 (Ser616), that is p-Drp1 (Ser616) expression was markedly (p < 0.01) enhanced in the hippocampal fraction of mice from the BCCAO group (Fig. 4A). EUX (30 and 60 mg/ml) administration in cerebral ischemia induced mice significantly (p < 0.01) upregulated the expression of Drp1 (Ser637) and markedly (p < 0.01) downregulated the Drp1 (Ser616) in the hippocampal specimen (Fig. 4B, C). The results suggest that EUX (60 mg/kg) treatment attenuates the mitochondrial fragmentation in cerebral ischemia induced mice.
Fig. 4
EUX suppressed hippocampal mitochondrial frag- mentation in BCCAO treated mice. EUX treatment in cerebral ischemia induced mice significantly upregulated the expression of Drp1 (Ser637) and markedly downreg- ulated the Drp1 (Ser616) expression in the hippocampal specimens. Data represent means ± S.E.M. (n = 8). Com- pared to the Sham group: #p < 0.05, ##p < 0.01, ###p < 0.001. Compared to the BCCAO group: *p < 0.05, **p < 0.01, ***p < 0.001
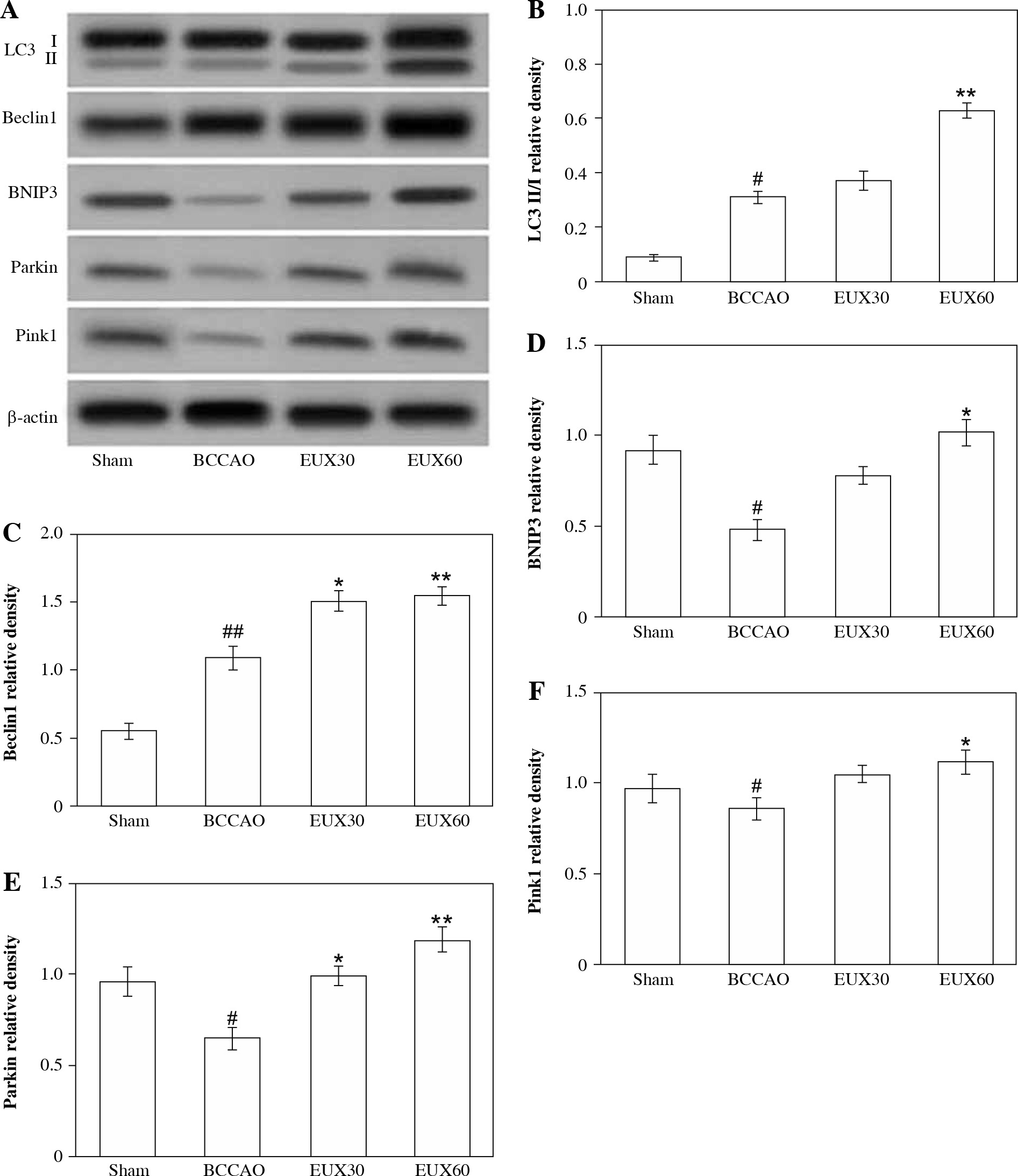
EUX enhanced hippocampal mitophagy in BCCAO treated mice
Compared to the animals from the Sham group, mice from the BCCAO group indicated a significantly (p < 0.01) higher LC3 II/I ratio and significantly (p < 0.01) increased expression of Beclin-1, which is an autophagic protein marker. The expression of LC3 II/I was significantly (p < 0.001) increased compared to Sham group animals. However, there was a significant (p < 0.01) reduction in the expression of LC3, Bnip3 and Parkin in the hippocampal fraction collected from mice from the BCCAO group. The hippocampal expression levels of LC3, Bnip3 and Parkin were markedly (p < 0.01) enhanced in the cerebral ischemic mice that received EUX (30 and 60 mg/kg), compared to the BCCAO group. EUX treatment moderately improved the levels of Pink1 after BCCAO treatment, compared to the Sham group (p < 0.05). The results suggest that EUX potentially triggers mitophagy in the hippocampus of the cerebral ischemia induced mouse model (Fig. 5A-F).
Fig. 5
Effect of EUX pre-treatment on the mitophagy sig- naling pathway proteins (LC3 II/I, Beclin1, BNIP3, Parkin and Pink1) in the ischemic mice using Western blotting. Data represent means ± S.E.M. (n = 8). Compared to the Sham group: #p < 0.05, ##p < 0.01, ###p < 0.001. Compared to the BCCAO group: *p < 0.05, **p < 0.01, ***p < 0.001
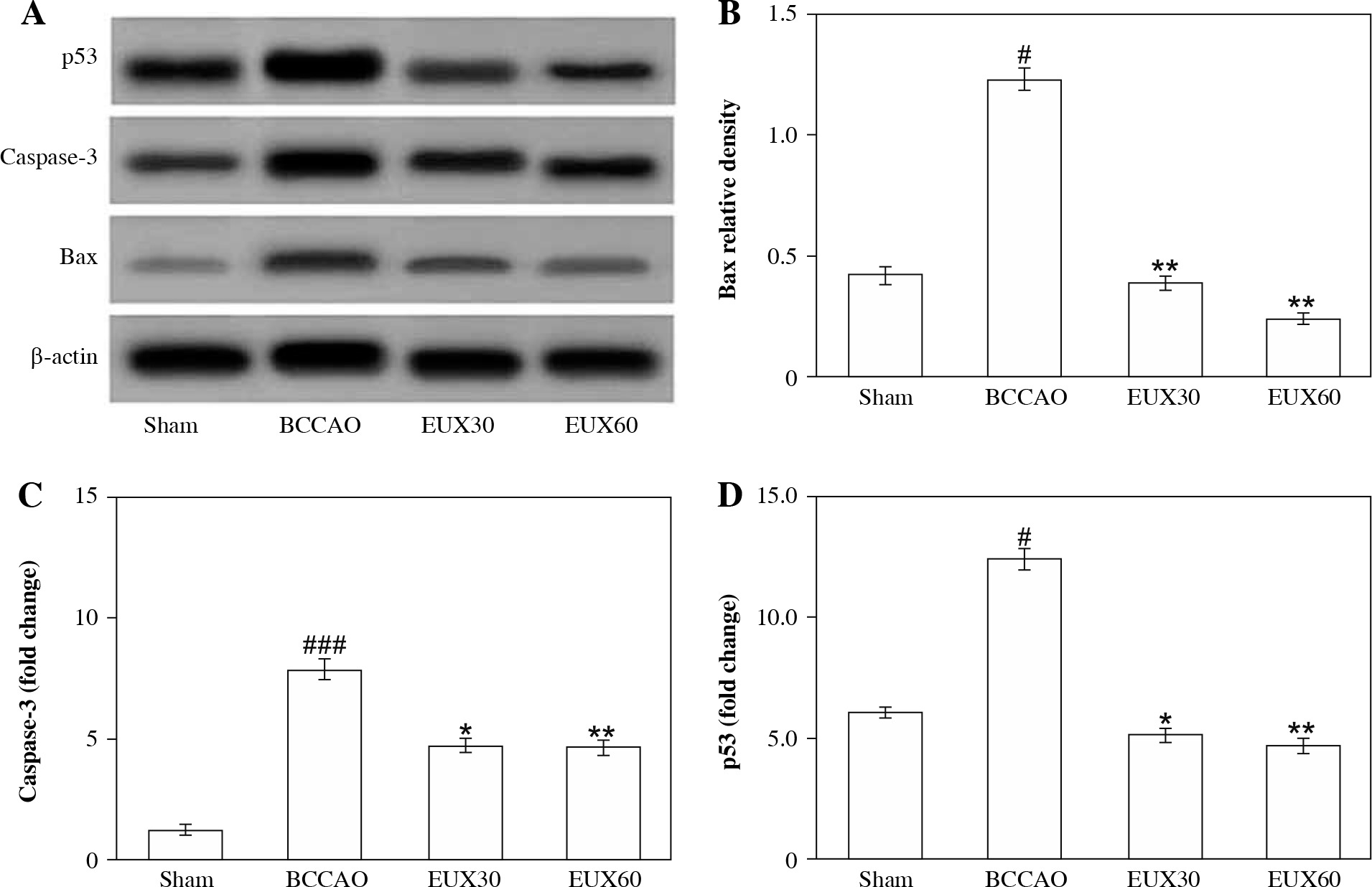
EUX suppressed apoptosis of hippocampal mitochondria in BCCAO mice
The immunohistochemical findings from the present investigation revealed that there was a remarkable (p < 0.001) rise in the hippocampal apoptotic protein caspase-3 expression after induction of cerebral ischemia, which was effectively (p < 0.01) normalized in hippocampus of mice treated with EUX (30 and 60 mg/kg) (Fig. 6A, B). An important player involved in the mitochondrial apoptotic pathway – Bax protein – was significantly (p < 0.001) increased after cerebral ischemia in the mouse model. EUX administration induced marked (p < 0.01) reduction of Bax expression in the hippocampus of cerebral ischemia induced mice (Fig. 6C). The expression of p53 was significantly (p < 0.05) increased in the mice from the BCCAO group, compared to the Sham group animals. However, EUX (30 and 60 mg/kg) administration significantly (p < 0.05) lowered the P53 expression, relative to the Sham group, to almost normal in the hippocampus of cerebral ischemic mice (Fig. 6D). The findings suggested that mitochondrial apoptosis regulated by p53 was targeted by EUX in the cerebral ischemic mouse model.
Discussion
Euxanthone, a xanthone derivative isolated from the plant Polygala caudata, has been known to exert several beneficial therapeutic effects. Our investigation revealed that treatment of cerebral ischemic mice with EUX attenuated the neurological deficiencies and corrected the cognitive deterioration caused by BCCAO. These protective effects were identified as the effect of EUX on mitophagy and apoptosis due to the oxidative burden coupled with mitochondrial breakdown during short-term cerebral ischemia and reperfusion injury. The results support promising use of EUX for such therapeutic indications.
Around 40% of human energy if expended by the brain tissues, which are highly susceptible to damage due to nutritional deficiency compared to other cells. The hippocampal region is responsible for learning and memorizing capability, and is primarily a sensitive target during cerebral ischemia. Hence, during our investigation the pathological alterations in the hippocampus were studied after induction of a cerebral ischemic event. Occlusion of the carotid artery results in partial cerebral ischemia and defective neurological function, resulting in ischemic episode and permanent vascular dementia [16]. Several investigations have indicated that a short-term drop in the cerebral perfusion may result in neuronal, motor and intellectual damage [9, 17].
Ischemia and oxygen deprivation results in ATP generation in mitochondria, which eventually leads to mitochondrial and respiratory cascade malfunction, leading to overproduction of free oxygen radicals that interact with cellular proteins and lipids and result in structural destruction of DNA. To determine the extent of degradation through oxidative stress mediated reactions, the MDA and SOD levels are measured [18]. The results of the present investigation indicated hippocampal oxidative damage after BCCAO treatment in mice, marked by significant increases in MDA and ROS levels, and a significant reduction in SOD level. EUX (30 and 60 mg/kg) administration normalized the MDA, ROS and SOD levels in the hippocampus of mice after BCCAO treatment, supporting the anti-oxidant potential of EUX after short-term cerebral ischemia coupled with reperfusion injury.
A critical balance between mitochondrial breakdown and integration is referred to as mitochondrial dynamics. In the event of oxidative stress, the mitochondrial membrane potential declines, which initiates the pathway involved in mitochondrial apoptosis and mitophagy. Drp1, a protein linked with mitochondrial kinetics, actively participates in the splitting of mitochondria, and levels of phosphorylated sites modulate the Drp1 activity. The Drp1 Ser 637 site undergoes phosphorylation by protein kinase A and is inactivated to a dormant stage in the cytoplasm [19]. PGAM5, a mitochondrial protein phosphatase, triggers dephosphorylation of Drp1 Ser 637, thereby resulting in Drp1 activation on the surface of mitochondria during stress conditions. This activated form of Drp1 triggers the process of mitochondrial splitting [20]. When conditions normalize, the Drp1 re-phosphorylates and reverts to the cytoplasm, increases mitochondrial survival and thereby balances the ATP generation within the cells [21]. In the present investigation, the p-Drp1 (Ser637) expression in its activated state was dramatically downregulated in the hippocampus of mice that underwent BCCAO treatment. On EUX (60 mg/kg) treatment, the expression of Drp1 (Ser637) was remarkedly increased. Under normal physiology, the Drp1 (Ser616) site is dephosphorylated and its expression influences Drp1 potential. During oxidative stress conditions, Drp1 (Ser616) undergoes phosphorylation and triggers mitochondrial breakdown. During cerebral ischemic conditions, neuroprotection is conferred when activation of Drp1 is inhibited [22]. The results of cerebral ischemia induced in mice by BCCAO during our investigation revealed an elevated level of p-Drp1 (Ser616). Stimulation of Drp-1 at Ser637 and Ser616 locations in mice subjected to BCCAO revealed an increase in the mitochondrial fragmentation during a combined short-term cerebral ischemia and reperfusion episode, which is inevitably associated with a resulting mitochondrial cascade leading to neuronal apoptosis. Treatment of cerebral ischemic mice with EUX (60 mg/kg) significantly expressed p-Drp1 (Ser637) and suppressed the Drp1 (Ser616) expression, supporting reduction in the mitochondrial breakdown in an event of cerebral ischemia.
Mitophagy is a process of selective removal of remains of damaged mitochondria, thereby reducing the mitochondrial membrane potential and arresting the cascade of mitochondrial apoptosis. Previous findings revealed that LC3 and autophagosomes gather in the proximal regions of the injury [9]. Bnip3 protein, a Bcl2 family member having a BH3 domain, is known to weakly influence apoptosis and is responsive to hypoxic and cerebral ischemic states. Its role is to link to the mitochondrial surface and promote distorted mitochondria to undergo autophagy. Bnip3, Parkin and Pink1 have also been identified as mitophagy linked proteins [23]. Though the involvement of Bnip3 in cerebral ischemia has not been identified previously, but there are investigations that revealed protection of myocardial cells mediated by Bnip3 through reduction of the cascade leading to mitochondrial apoptosis [23]. ATP and ROS production is propelled by Pink1 and Parkin proteins, which further modulate fission and fusion of mitochondria [23]. Deficiency of Pink1 and Parkin in neurons results in Drp1 accumulation, which eventually leads to exaggerated fission and thereby enhanced oxidative stress and lowering of ATP generation. A proper balance is required between formation of new mitochondria and mitophagy for removal of mitochondria that have undergone depolarization and distortion, but under stress conditions any disturbance in the phenomenon results in cell death [24]. Pink1 may attenuate non-mitochondrial oxidative stress. Parkin blocks the mitochondrial permeability transition pore (PTP), which imparts neuronal protection from apoptotic cell death that is initiated by the mitochondrial cytochrome c release. The Pink1-Parkin cascade is involved in maintaining a delicate balance between mitochondrial fission and fusion, lowers oxidative stress in mitochondria, impedes fission, and retains membrane potential of the mitochondria [23]. Previous investigations suggest that reducing fission or enhancing fusion, or specifically regulating mitophagy, may be used as an effective strategy to treat neurodegenerative disorders [8]. Decline of membrane potential of mitochondria results in translocation of Pink1 into damaged mitochondria, which subsequently mobilizes their inherent catalytic capacity to favor autophagy [8]. In the present investigation, LC3 and beclin1 expression triggered autophagy, which was stimulated after cerebral ischemia, and the autophagic potential was elevated after EUX treatment. Protein Bnip3 expression arbitrated through autophagy was significantly enhanced after EUX treatment, and this revealed that the extent of autophagy was elevated after cerebral ischemia and reperfusion. The Parkin protein expression was enhanced after inducing cerebral ischemia, but Pink1 expression was not altered significantly. The mitophagic activity was enhanced after EUX administration, indicated by a significant rise in Perkin expression and a moderate rise in Pink3 expression. These outcomes suggest that mitophagy regulated by the Pink1/Parkin cascade actively affects the pathogenesis of cerebral ischemia, and EUX induces therapeutic activity by concurrent elevation in mitophagy and autophagy, thereby conferring neuroprotection after short-term cerebral ischemia and reperfusion.
Previous investigations reveal that Bax triggers mitochondrial apoptosis mediated through inculcation of PGAM5 (phosphoglycerate mutase 5)-Drp1 complexes over the mitochondrial surface [25]. An elevation in the expression of caspase-3, a prominent marker protein in the apoptotic cascade, indicates the manifestation of the apoptotic process. Findings of our investigations revealed that P53, caspase 3 and Bax protein expression levels in hippocampal tissues of mice were significantly enhanced in the BCCAO mouse model, whereas the expression of such apoptotic-linked proteins was remarkably downregulated after EUX treatment.
Taken together, the effect of EUX can be attributed to normalization of oxidative stress markers in the hippocampi of the affected mice. The autophagic potential was enhanced after EUX treatment, as revealed by LC3 and Beclin1 expression in the hippocampus. Expression of Bnip3, Pink and Parkin1 was elevated after EUX administration, indicating mitophagy caused by EUX after ischemia and reperfusion in mice. Increased expression of Bax, p53 and caspase-3 expression after an ischemic episode were corrected after EUX treatment, indicating a neuroprotective effect. The findings thus support the neuroprotective effect of EUX in a cerebral ischemia and reperfusion model induced in mice.