

Introduction
Severe infections are a major public health problem responsible for about 40% of hospitalizations in intensive care units (ICU). Despite latest treatment methods and a decrease of sepsis-related deaths during recent years, mortality rate is still high, corresponding to 46% in Poland [1]. The United States incidence of sepsis is 0.3%, and is responsible for 63.7% of hospitalizations in ICU, with mortality rate of 30%. [2, 3]. Whereas in case of severe sepsis and septic shock, the mortality level is as high as 25% and 50%, respectively. In Great Britain, 30,000 cases of severe sepsis are diagnosed each year, with mortality rate of 35%. The Germany national incidence of sepsis increased by 15% within 2007-2013 and in 2013, 314 cases were diagnosed per 100,000 inhabitants. However, mortality rate of 30% has been recorded for years. Severe sepsis incidence was 173/100,000, with mortality rate of 46% [4]. According to these authors, epidemiological studies regarding sepsis demonstrated varied results, due to some difficulties regarding diagnostics of severe infection and progression of the disease as well as the absence of clear guidelines. In terms of an attempt to provide practical solution, the Society for Critical Care Medicine (SCCM) established modern guidelines regarding the third international definition of sepsis and septic shock [5] that amended the traditional definition of severe infection [6]. According to the new definition, the sepsis is a deregulated immune response to an infection that leads to organ dysfunction (both immunological and non-immunological) that poses hazard for human life, whereas septic shock is a life-threatening complication of sepsis that leads to dangerously low blood pressure and abnormalities in cellular metabolism, which often results in death. The new classification removed a definition of severe sepsis. Also, the approach regarding assessment of systemic inflammatory reaction syndrome (SIRS) was modified as the potentially first phase of sepsis. At present, sepsis is considered as a simultaneous triggering of pro-inflammatory and anti-inflammatory processes as well as further disturbances of immune system, interruptions of cardio-vascular system, nervous system, endocrine system, clotting, bio-energy, and cellular metabolic pathways. These disturbances cause dysfunction of organs, therefore the definition of severe sepsis was no longer required [5].
High mortality of patients diagnosed with severe infection is caused by largely unknown mechanisms of sepsis-induced immune system response. Initially, an assumption was made that the main therapeutic difficulty in case of sepsis was associated to bacteremia and organism incapacity to fight back the infection. However, thanks to the development of research in immunology and genetics, the key difficulty regarding severe infection treatment is related to pathology of very dynamic and varied immune system response to a massive infection, e.g. SIRS, compensatory anti-inflammatory response syndrome (CARS), or persistent inflammation and immune suppression catabolism syndrome (PICS) as well as organ-related effects of this reaction [7, 8]. It turned out that in case of patients diagnosed with post-injury severe infections, the short-time parallelly trigger over 5,000 genes responsible for pro-inflammatory and anti-inflammatory response as well as growing immune suppression and protein-energy malnutrition, related to the biggest challenges of contemporary therapy in severe infections [9-13]. According to these studies, in case of ICU septic patients with high-risk of mortality, the solutions would require mainly diagnostics of early immune and genetics (e.g. determining prognostic markers for infection) and biological treatment according to the phase of immune response.
Selected signaling pathways of Toll-like receptors
Mechanisms responsible for severe infection-related immune system response, attract an extensive interest, in particular, of non-specific defense mechanisms including signaling pathways of Toll-like receptors (mainly TLR4 and TLR2) that recognize distinct pathogen-associated molecular patterns (PAMP) and play a critical role in innate immune response. Studies conducted within last three decades confirmed a significant role of TLR signaling pathway in case of tumors, inflammatory responses, and severe infections. Identification of TLR receptor ligands (external and intra-cellular) and better understanding of proteins responsible for signal transduction enable the possibility of inhibition and signals blocking at different phases of cell activation, as these action may have significant role in terms of therapy. The first attempts related to such therapy were promising, but later clinical tests with patients suffering from severe infection did not reveal significant reduction of mortality rate [14]. However, these studies confirmed poor knowledge and high complexity of pathomechanism behind sepsis-induced innate antibacterial response disorder. In addition to a standard therapy administered to these patients, it was difficult to determine whether biological treatment (anti-inflammatory, immunity-stimulating, limiting lymphocyte apoptosis) or “aggressive” nutritional treatment compensation of protein-energy deficit (immunonutrition and pharmaconutrition), impacted the decrease of sepsis-related mortality [13]. In order to better understand the diagnostic and treatment issues related to high mortality of patients with severe infection, a summary is necessary concerning the-state-of-the-art of innate anti-microbial resistance mechanisms associated with signaling pathways of selected Toll-like receptors.
There are ten TLR identified in humans. TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are situated in the cell membrane, whereas TLR3, TLR7, TLR8, and TLR9 are located in endosome and lysosome membrane. TLR receptors recognizing different PAMPs are expressed mainly in epithelial cells and leukocytes. Their role relates to limitation of infection, due to release of cytokines, nitrogen monoxide, local phagocytosis activation, and stimulation of adaptive immune system. TLR and retinoic acid-inducible gene-I-like receptors (RLR) signaling pathways (recognizing virus RNA) and nucleotide-binding oligomerization domain-like receptor (NLR), which could recognize intracellular patters including NOD1 and NOD2 receptors, create a connection, which provide a combination of innate and adaptive immune response as effectors of pathways stimulating lymphocytes. Studies confirmed that TLR2 receptors are susceptible to ligands from Gram-positive bacteria, whereas Gram-negative bacteria patterns produce TLR4 receptor ligands. TLRs receptor ligands and NOD1/NOD2 are provided with varied PAMPs that, in different cases, may be supplied by one bacteria. In case of NOD1, it is γ-D-glutamyl-meso-diaminopimelic acid that delivers ligand, which is characteristic for Gram-negative bacteria and specific Gram-positive bacteria. The main role of NOD2 includes recognizing muramyl dipeptide acid (MDP) being a component of bacteria cell wall. In case of TLR2, ligands are provided with inter alia lipoteichoic acid and lipoproteins, whereas lipopolysaccharide produces for bacterial ligand for TLR4 [15]. It was proven that reduction of TLR2 receptor expression in human monocyte cells resulted in increased tolerance to LPS. However, reduced activity of TLR2 receptor resulted in increase of sepsis-related mortality [16]. Studies conducted by Bergt demonstrated that mice with TLR2 deficit showed longer survival when under severe infection [17]. Some studies were also conducted on mice with TLR2 and TLR4 antibodies, which indicated reduced infection after application of receptor antagonists in case of sepsis caused by Gram-negative and Gram-positive bacteria [18, 19]. Moreover, these studies indicated that deficit of TLR4 increased susceptibility of mice to sepsis caused by Gram-negative bacteria, yet prevented sepsis caused by endotoxins [20]. Similarly, a deficit of TLR2 increased susceptibility to diseases caused by Gram-positive bacteria [21]. Depending of bacteria causing infection (transporting specific ligands), an activation of varied or numerous receptors takes place, which initiates a problem related to use of antagonist of these receptors without prior culture. Of note, modulation of signaling pathways is crucial at the early phase of infection [22].
Studies indicated that TLR 2 and 4 receptors related to membrane glycoprotein embedded in a cell, which enabled reading of environment signals that trigger signaling cascades. Finally, cell nucleus activation and induction of gene expression coding pro- and anti-inflammatory cytokines occur. Receptor structure consists of two parts: 1) an internal cytoplasmic part of TLR, C-terminal domain TIR (Toll-IL-1-receptor), named due to high homology to 1 type 1 interleukin receptor (IL-1R1), and 2) N-terminal domain of TLR, named LRR, which is built from leucine-rich repeats. TIR domain is crucial for the activation of cascade signal, as a result of contact with adaptor molecules. The purpose of conducted studies was inhibiting of TIR domain and consequently, the whole TLR4. However, clinical tests did not confirm the efficiency of such therapy for sepsis-related cases [23, 24].
Essential elements of studies regarding the innate antimicrobial response disorder are adaptor proteins that are significant regarding diagnostics and therapy in case of patients with severe infection. Adaptor proteins are selectively stimulated by TLR4 receptors and include myeloid differentiation primary response (88MyD88), TIR domain containing adaptor protein (TIRAP), TIR-domain-containing adapter-inducing IFN (TRIF), and (TRIF-related adaptor molecule (TRAM). There are MyD88-dependent and MyD88-independent (TRIF-dependent) pathways that vary according to adaptor protein carrying the signal to IL-1receptor-associated kinas-4 (IRAK4). As a result of indirect or direct binding with MyD88 or TRIF adapter molecule to TLR, activation of kinas IRAK-4 follows. Consequently, phosphorylation is provided to IRAK-1 protein. Activated kinas of IRAK-1 released to cytoplasm is connected to TNF-receptor-associated factor (TRAF6). Formation of complex results in activation TAK1-binding protein (TAK1). The activated complex TAK1 stimulates IKK enzyme (inhibitor of IκB kinas) that causes phosphorylation and degradation of NF-κB inhibitor (nuclear factor κB), namely IκB protein (inhibitor of kappa B). Next, as a result of IκB inactivation, a release of NF-κB transcription factor occur that penetrates the cell nuclei and induces an expression of genes coding the pro-inflammatory cytokines [25-31]. The excessive stimulation of receptors and excessive production of inflammatory cytokines result in strong inflammatory reaction within whole system. Figure 1 describes TLR2 and TLR4 pathways with antagonists.
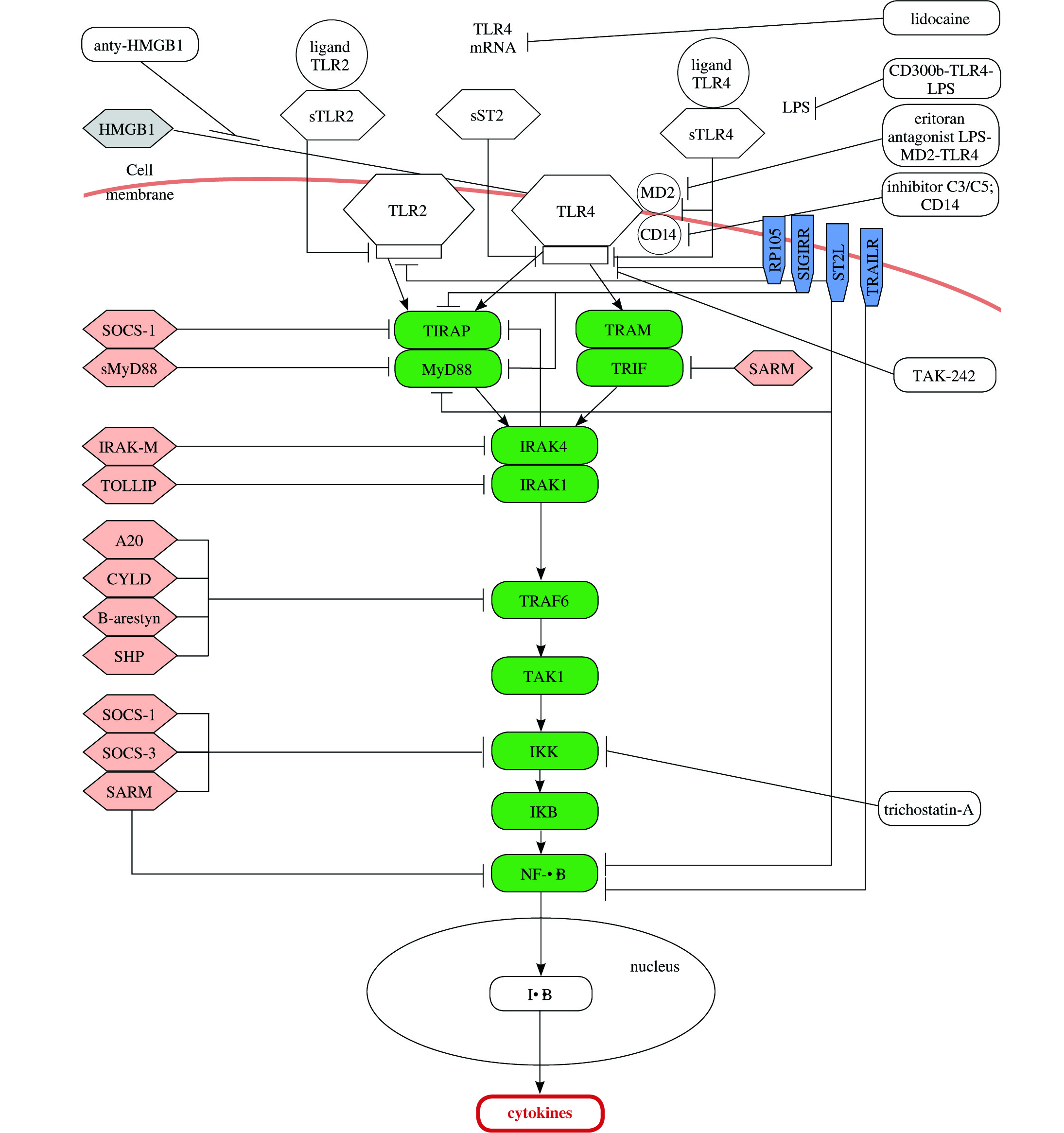
The primary cause of severe infection related to strong inflammatory reaction in still unknown. Some signaling pathways were identified associated with innate immune system that may be responsible for the origin of primary cause. Sepsis is caused by endotoxins that react with patient cells (leukocytes, epithelial cells) through specific receptors of cell surface and trigger signaling pathways that stimulate potentially deregulated immune response. Consequential disturbances may follow inside the cells with regard to the cell cycle (e.g. chromosome condensation disturbance, DNA replication disturbance [32], apoptosis [33]) or with regard to immune response. Activation or modification of signal transduction occur within the following pathways: heat-shock protein 60 (HSP60) [34], heat-shock protein 70 (HSP70)/TLR [35], TLR2, TLR4, high mobility group box 1 protein (HMGB1)/TLR) [36], IL-1 pathway [37], platelet-derived growth factor (PDGF) via signal transducers and activators of transcription (STATs) [38]. Modifications will also follow with regard to dendritic cells [39] and activation e.g. natural killer T (NKT) cells [40] as well as cell cycle control or apoptosis [41]. Further problem that deteriorate patient condition relates to expression reduction of chloride channels formed by cystic fibrosis trans-membrane conductance regulator (CFTR), as a consequence of activation of TLR4 receptors and a release of NF-κB. This leads to dysfunction of respiratory tract epithelial cells and the progression of lung inflammation (caused by neutrophils infiltration and locally produced inflammatory mediators triggering vascular endothelium injury), which results in high-risk of lung failure [42, 43].
From the clinical perspective, it was TLR4 that was responsible for the most of above specified pathways. It turned out that during the first phase of inflammatory response, the stimulated TLR pathway activated NK-κB to a degree that caused simultaneous ejection of pro- and anti-inflammatory cytokines. Initially, pro-inflammatory cytokines are dominant, whereas during further phase of disease, cells activation cause an intense production of anti-inflammatory cytokines, of which concentration corresponds to dominating values [44, 45]. Some studies demonstrated that during a severe infection, the progression of disease results in expression of intensity modulation of particular genes within TLR pathway (TLR2, TLR4, CD14, NF-κB pathway) [46]. Also, various expressions were confirmed with genes within PBMC (peripheral blood mononuclear cells) and neutrophils. In case of PBMC, the progression of the disease resulted in reduction of gene expression within TLR pathway [47]. Neutrophils demonstrated a different pattern of adaptation, as the progression of the disease in this case resulted in increased gene expression [47]. In addition, other dysfunction was evident during sepsis with regards to neutrophils, e.g. incapability for chemotaxis. Usually, these cells are subjected to an apoptosis within 24 hours following releasing from bone marrow, however during sepsis, their life cycle extends. Similarly, massive release of immature neutrophils from bone marrow occurs, resulting in significant increase of circulating neutrophils of different maturity [48]. This phenomenon lead to multiplied gene expression of TLR4 pathway and subsequently increased production of effectors (pro- and anti-inflammatory cytokines). Surprisingly, different behavior is presented by lymphocytes, which demonstrate accelerated apoptosis [49-51]. These facts additionally highlights the dominant role of the innate immune response during severe infection.
Studies conducted on mice proved that absence of any TLR, NOD receptor or adaptor protein did not prevent immune response provided by other TLR and NOD receptors and their pathways [15, 52, 53]. On the other hand, available data suggests existing redundancy (synergism) between MyD88- and TRIF-dependent pathways that increase activation of receptors. Studies conducted on mice showed also that innate deficit of MyD88 and TRIF genes resulted in increased survival during severe infection, due to efficient reduction in the number of bacteria within peritoneum and peripheral blood, as a consequence of efficient antibiotic treatment. Additionally, the absence of MyD88/TRIF signaling prevented the excessive production of IL-10 [54]. On basis of this information, one can speculate that the control over specific TLR pathways provided during sepsis with regard to adaptor protein, may ensure a new therapeutic strategy [53].
Signaling pathway modulation
There are two possible options regarding modulation of TLR signaling pathway that may result in blocking ligand binding with TLR or modulation of intracellular signaling pathways to inhibit signal transduction. Towards a better understanding, the immune response control mechanisms, and different therapeutic measures were developed to inhibit TLR-dependent signaling and to control severe infection. Inhibiting factors are classified as: low-molecular inhibitors (TAK-242) [24], anti-bodies [55], oligonucleotides, A-lipid analogue (Eritoran) [14], microRNA (miR-21) [56], and recently discovered nano-inhibitors (glycolipid-coated GNP [57], NAHNP [58,59], HDL-like NP [60]) [61]. Most recently developed inhibitor lidocaine protects kidneys and liver during sepsis, due to reduction of TLR4 receptor expression within these organs’ tissues. Administration of lidocaine during studies prevented these organs against injury or failure [62]. Previous studies indicated that administration of lidocaine reduced chemotaxis in lungs, which reduced neutrophils infiltration and prevented very common complications during severe infection – lung failure [63].
Many proteins were discovered as the inhibitors for TLR2 and TLR4 signaling pathway of particular adaptor protein that had a significant role with regard to treatment of severe infection. Studies indicated that inhibiting of signaling pathway may occur in practice at every phase, both in intra-cellular and extra-cellular manner [64]. The best understood mechanisms of TLR receptor signaling negative control in immune cells, where signaling inhibition occurs as early as on cell surface is the ability of surface receptor inhibition demonstrated by TLR – sTLR (soluble TLR) that are appropriate for each receptor – sTLR2 and sTLR4 [65]. Other possibility regarding inhibition of TLR4 pathway activation relates to antibodies of this receptor. At present, clinical tests are focusing on NI-0101 antibody that is the first monoclonal antibody (mAb) blocking TLR4 signaling, and prevents the release of cytokines upon LPS ex vivo and in vivo administration and increase of C-reactive protein (CRP) as well as flu-like symptoms expected after LPS in vivo administration [55]. Other option regarding signaling pathway modulation is related to membrane protein sharing a TLR domain, with TIR domain, namely ST2L proteins (ST2-ligand) [66], SIGIRR (single immunoglobulin IL-1-receptor-related molecule), RP105 (radioprotective 105), and TRAIL-R (tumor necrosis factor (TNF)-related apoptosis inducing ligand). The further phases of TLR4 signaling pathway provided possibility regarding signal transduction stoppage by cytoplasmic inhibitors, which controlled performance of particular adaptor protein and enzymes. These inhibitors included: SOCS (suppressor of cytokine signaling), MyD88s (short myeloid differentiation primary response protein), SARM (sterile α and HEAT-Armadillo motif proteins), IRAK-M (IL-1R-associated kinas-M), TOLLIP (Toll-interacting protein), A20 (TNFAIP3 – tumor necrosis factor–induced protein 3), CYLD (cylindromatosis), β-aresyn, SHP (small heterodimer partner) [64]. Modulation of TLR4 signaling pathway is also possible with HMGB1 protein antibody [67]. This protein was released as a result of activation of immune cells (monocytes, dendritic cells, macrophages). HMGB1 fulfils the role of cytokine during inflammatory response and activates TLR4 signaling pathway, also when no infection followed. Studies demonstrated that during sepsis, concentration of HMGB1 increased suddenly, which resulted in further intensified activation of TLR4 signaling pathway. This adverse process may be suppressed by anti-HMGB1 antibodies. Studies proved that application of anti-HMGB1 antibodies protected against organ failure during sepsis [67]. Researches on mice showed that reduction of HMGB1 concentration responsible for caspase-1 activation correlated with immunity to LPS [68]. Previous studies focused on other proteins responsible for modulating of TLR4 receptor functions such as antagonist-LPS-MD2-TLR4 (Eritoran) [14], TLR4 antibodies, inhibitor proteins of TLR4, e.g. TAK-242; however, there was no reduction in the number of deaths in patients treated with the above mentioned [23]. Nevertheless, very promising seemed to be proteins blocking MyD88, since a reduced intensity of response was demonstrated to staphylococcus enterotoxins, following blocking of this important adaptor protein, which is common for TLR2 and 4 signaling pathways [69-71]. Also promising were results of studies regarding trichostatyn A (TSA), which suppresses MyD88-dependent TLR4 signaling. To examine an impact of TSA on signaling within TLR4 and TLR2, studies were conducted regarding the expression of selected proteins within these pathways. Researches were performed on mice that included cecal ligation and puncture. A reduction was observed regarding TLR2, TLR4, and MyD88 concentration in mice with induced TSA comparing to those without TSA before induction. At the same time, TSA samples did not demonstrate TRIF protein concentration reduction [72]. Suppression of MyD88-dependent signaling, upon TSA administration, resulted in inhibition of TNF-α and IL-6 concentration increase in serum taken 6 and 24 hours after treatment. Moreover, mortality reduction was observed [72]. As many attempts may have failed regarding selective modulating of adaptor proteins active and a release reduction of effectors for TLR4 signaling, a new method of inhibition was proposed. The next concept concerning modulating of TLR2 and TLR 4 signaling pathway was associated with concurrent suppression of cells containing TLR receptors at two phases of their stimulation. Actually, the inhibition was proposed on so called ”narrow neck” before activation of receptors, by way of inhibition of central components of complement system–C3 or C5 proteins as well as neutralization of CD14 cell being coreceptor for TLR4 and TLR2. The purpose of C3 and C5 neutralization was the reduction of general inflammation, owing to limited recognition of PAMPs (pathogen-associated molecular patterns). This theory provided good results during in vitro tests, both in case of infection-induced inflammatory reactions and non-infectious ones [73]. Control over “narrow-neck” cells of immune systems’ two elements (of complement system and TLR signaling pathways) and prevention of PAMP and DAMP (damage-associated molecular pattern) may provide very efficient method for weakening of inflammatory response, as well known innate immune systems that support each other. The double inhibition may provide general, comprehensive therapy pattern against many diseases, when innate immunity is activated in wrong manner [73]. Other possible modulation tested during in vitro tests related to CD300b receptor, which provided LPS binding along with TLR4. CD300b-TLR4 complexes controlled both MyD88 and TRIF-dependent (MyD88-independent) signaling pathways. Complex CD300b-TLR4-LPS stimulated pro-inflammatory response and reduced anti-inflammatory response, which prevented injury of tissues during the course of severe infection [74].
In spite of research indicating the links between TLR2 and TLR4, and TLR and NOD signaling pathways, in response to bacterial infection as well as many attempts regarding modulation of signaling pathways, disorders regarding their control require further knowledge. Attempts concerning modulation of signaling pathways did not contribute to reduction of high mortality at ICU, although only an immediate therapy provides opportunity for modulation or inhibition of early antibacterial response, and may protect the patient against the results of excessive activation of immune response. In case of severe infection, an appropriate biomarker should be helpful in early detection and further assessment of progression of the disease, treatment monitoring, and further prognosis in relation to critical complications or death.
Selected sepsis biomarkers
Until now, research covered over 170 biomarkers of sepsis [3, 75], which indicated the problem related to complex mechanisms of severe infection. Some biomarkers of sepsis as well as their application for diagnostics and forecast purposes were described in Biron et al. work [76].Most of studied factors did not find application in clinical practice, mainly due to absence of impact on mortality [77]. Table 1 presents the most popular markers.
Table 1
Potential sepsis diagnostic biomarkers
[i] ICAM-1 – intercellular adhesion molecule 1, CD – cluster of differentiation, sTRE M-1 – soluble triggering receptor expressed on myeloid cells-1, MIF – macrophage migration inhibitory factor, Gas6 – growth arrest-specific 6, sTNFR – soluble tumor necrosis factor receptors, aPTT – activated partial thromboplastin time, miR – micro RNA
Researches regarding new and more efficient diagnostics biomarkers were mostly focused on indicators related to innate response to infection as well as connections of pro-inflammatory response with anti-inflammatory response [78]. To improve results of treatment and survival, doctors needed reliable tools for precise clinical assessment while facing difficult challenges, e.g. differentiation of sepsis from SIRS or assessment regarding the phase of immune response. The significance of diagnostics and determining the phase of response to infection are essential for scheduling safe and effective therapy as well as administration of appropriate nutritional treatment. In fact, sepsis mediators such as cytokines attracted particular attention.
Cytokines
Cytokine production activation in the course of severe infection is connected with liposaccharide binding (LPS) to TLR-4, causing monocytes to product cytokines that are crucial for inflammatory response, which is evident during sepsis or after multi-organ trauma. Research regarding using some cytokines as sepsis biomarkers were conducted for many years. It was proven that cytokines were not only the effectors of signaling pathways responsible for the response of infection, but were engaged in controlling immunological response. The conducted experimental studies involved an assessment of TLR-4 expression within isolated monocytes in the presence of tumor necrosis factor (TNF) -α, interleukin (IL) 6, IL-8, and IL-10 as well as activation of cells during administration of treatment. TNF-α significantly reduced expression of mRNA TLR-4 after 6 hours, consequently leading to a reduction of TLR-4 surface protein expression depending on dose and time. After 48 hours from the TNF-α treatment, the reduced translocation of nuclear factor-κB was found with reduced release of IL-6, followed by LPS stimulation. Moreover, incubation of monocytes with IL-6 increased the level of TLR-4 cell surface protein and improved the potential for activation of NF-κB upon LPS stimulation. The stimulation of monocytes with IL-8 or IL-10 did not have significant impact on NF-κB activation. Studies indicated that not only LPS, but also TNF-α and IL-6, could control immune response through TLR-4. In our opinion, the reduced activation of TLR-4 by TNF-α associated with LPS hiporeactivity during formation of NF-κB, whereas the increased activity of TLR-4 with IL-6 may have increased the reaction of mononuclear phagocytes [79]. As early as the first hours following the injury or infection, an increase of TNF-α, IL-6, and IL-1β concentration in blood was observed [80,81]. TNF-α is one of the main two mediators for immune response control and marker of severe infection-related pathophysiological changes. During the early phase of infection, it was possible to determine the maximum concentration of cytokines including TNF-α, IL-1, IL-6, IL-8, and IL-10. Then, an excessive inflammatory response concurrently follows the anti-inflammatory response, frequently leading to immunosuppression [79]. The deregulated expression of cytokines in the course of proinflammatory response increase of IL-6, TNFα, IL-1, and IL-8 concentration, resulted in an increased mortality, and no therapy against a single cytokine carried significant benefits other than already obtained during standard procedures [82]. The correlation was observed between high concentration of IL-6 and mortality risk [83]. In case of TNF-α and IL-1β, a research on applied antagonist did not demonstrate any prognostic value. However, a correlation was indicated between high concentration of IL-8 and the risk of early death (within 28 days) [84]. A study by Opal et al. indicated that IL-1Ra was a strong mortality predictive indicator during sepsis [85]. However, results of a research by Cioara et al. did not confirm any correlation between the increase of IL-1Ra concentration and mortality rate during severe infection, possibly due to small group of patients [86]. In spite of many studies on the release of cytokines during inflammatory response and in the course of sepsis, the interpretation of results was still difficult, and it was hard to specify physiological and pathological response and to establish some standards [75]. Cytokines (TNF-α, IL-1β, and IL-6) were unspecific in case of sepsis, and were more useful as prognostic markers than diagnostic ones [87]. The exception related to IL-6 of proven diagnostic value was demonstrated as a correlation with concentration of PCT and CRP [88].
Other essential element of sepsis was CARS in immunosuppression, for which IL-4 and IL-10 played a significant role. IL-4 is a cytokine produced by basophiles, mastocytes, and lymphocytes, classified as an early marker of CARS. Along with IL-10, it inhibits synthesis of pro-inflammatory cytokines in cells stimulated with LPS [89]. IL-10 is an anti-inflammatory cytokine, playing a significant role in development of CARS. IL-10 is produced by CD4+ Th2 cells, monocytes, and B lymphocytes. Concentration of IL-10 in patients with diagnosed sepsis was slightly higher, resulted in immunosuppression and worse prognosis and mortality [84,90,91]. However, this marker was presented with low sensitivity and specificity that prevents its clinical use.
Sepsis relates to a disease including many disorders, different outcomes as well as different genetic disorders and immune response. In spite of conducted studies, the determination of cytokine concentration for sepsis monitoring is not a standard procedure, although is recommended by many anesthesiologic societies.
Cluster of differentiation-64 – a marker of early immune response
Regarding searching for sepsis new biomarkers, very promising studies were performed on monocytes with cluster of differentiation-64 (CD64) receptor expression. CD64 is a IgG receptor with high affinity, constitutively present in monocytes and in small amount in polymorphonuclear cells (PMN). In case of infection, the expression of CD64 on PMN increases significantly to provide more phagocytosis. On account of that, CD64 possesses a few desired features specific for biomarkers, and may be used to differentiate bacterial infections from other types of inflammation without infection. Its expression upon LPS activation is slight in case of neutrophilia in septic patients and drops highly upon activation within a few hours [92].
It was proven that CD64 expression on polymorphonuclear cells taken from healthy people, increased within 20 minutes to 2 hours upon induction of lipoteichoic acid or lipopolisaccharide, and maintained for 48 hours. Within few following days, its disappearance standard level of CD64 expression was restored [93]. Moreover, CD64 is relatively stable after blood collection, and measurement of its expression is easy with flow cytometry. The increase of CD64 concentration on PMN was indicated in patients with diagnosed severe infections [94]. For septic patients, the expression changed as a result of antibiotic therapy as well as CD64 markers changed due to antibiotic therapy [95]. Note that CD64 over-expression in blood monocytes and neutrophils in patients with septic infection seemed to be connected with dysfunction of leukocytes comparing to CD64-negative cells [96]. The meta-analysis of 13 studies including children and adults indicated that CD64 expression on PMN may be used as a diagnostic parameter for bacterial infection for 79% (95% CI: 70-86%) and 91% (95% CI: 85-95%) of sensitivity and specificity, respectively. The prospective studies performed on babies and children indicated that CD64 expression demonstrated higher diagnostics precision in case of severe infection, comparing to PCT and CRP [97].
Triggering receptor expressed on myeloid cells-1
Out of potential biomarkers of sepsis, triggering receptor expressed on myeloid cells (TREM) signaling pathway protein was also considered. TREM-1 is glycoprotein, which is present on the surface of neutrophils, mature monocytes, and macrophages. It was proven that protein is engaged in signaling of inflammatory immune response during infection. A soluble counterpart TREM-1 (sTREM-1) was identified in biological liquids of patients and animals with diagnosed different infections and diseases, and its correlation was often evident with a disease progression. According to experimental studies, TREM-1 had a pathogenic effect on organ dysfunction, and suppression of TREM-1 pathway was beneficial in case of Streptococcus pyogenes-induced sepsis [98]. Prospective clinical studies including newly admitted critically ill patients with suspected infection, covering 3 biomarkers TREM-1, PCT, and CRP, showed that measurement of plasma levels of soluble TREM-1 could help to rapidly identify those patients with infection. [99]. Some perspective studies with patients diagnosed with severe sepsis and septic shock demonstrated a significantly higher concentration of sTREM-1 in plasma of patients who did not survive, during admission at ICU, comparing to patients who survived. Moreover, the level of sTREM-1 in plasma remained significantly high until death, comparing with patients who survived [100]. However, these data were not consistent in clinical studies. In a similar study, Gibot et al. confirmed that level of sTREM-1 was significantly lower in patients who died, comparing to those who survived. Nevertheless, in case of low level during admission, the level of sTREM-1 was stable and slightly high in patients who died, with reduced number in those who survived [101]. Available data indicated that TREM demonstrated some features of predictive marker, although it did not meet the requirements regarding diagnostic marker. Studies in patients after cardiac surgery and cardiac arrest without infection indicated that sTREM-1 and PCT were not specific for infection and their possible increase during severe inflammation without infection to the degree observed in patients diagnosed with sepsis [102]. Moreover, the meta-analysis performed in 2012 covering eleven studies indicated that sTREM-1 in plasma determined individually was not a sufficient biomarker differentiating sepsis from SIRS [103]. In 2016, a cohort study indicated predictive value of sTREM-1 protein as an indicator for sepsis occurrence in patients suffering from polytrauma [104].
High mobility group box 1 protein
High mobility group box 1 protein (HMGB1) protein is essential in immune inflammatory response to infection and trauma. The main receptors of HMGB1 protein are RAGE receptors (receptors for advanced glycation end products) and TLR receptors included in immune system cells and endothelial cells. Although both receptor groups had different signaling pathways, they finally activate NF-κB, which trigger gene expression of cell adhesion molecules, pro-inflammatory cytokines, and proangiogenic factors. HMGB1 protein is crucial for pro-inflammatory response, inter alia by macrophages activation for production of pro-inflammatory cytokines, activation of enterocytes for release of nitric oxide, and endothelial cells activation for release of pro-inflammatory cytokines, chemokines, and adhesion molecules [105]. In intracellular environment, HMGB1 protein is localized mainly in cell nucleus, where it is involved in replication, recombination, transcription, and repairing of DNA [106]. Release of HMGB1 nucleus may follow to extracellular compartment through, e.g. dead of living cells, in response to bacterial exogenous endotoxins and pro-inflammatory cytokines [107]. When in the extracellular compartment HMGB1 becomes pro-inflammatory cytokine to activate inflammation and cell proliferation, it becomes a chemotactic factor, stimulating angiogenesis, activating endothelial cells for the release of IL-8 and TNF-α. HMGB1 protein also involved in regeneration of damaged tissues [108]. Studies indicated a high level of HMGB1 in patients suffering from injuries, in particular acute inflammation, including SIRS [109]. High concentration of this protein resulted in worse prognosis during severe infection, when there was an evident correlation with HMGB1 high concentration and mortality rate. It was proven that application of anti-HMGB1 antibodies resulted in reduced pro-inflammatory response without an increase of immunosuppression, lower mortality rate, and better prognosis. High concentration of HMGB1 leads to immunosuppression [82]. Studies on mice confirmed long-term increase of oxygen concentration that resulted in an increase of extra-cellular HMGB1 concentration within respiratory tract in animals that underwent hyperoxia. This condition resulted in dysfunction of macrophages and consequently ventilator-associated pneumonia. At the present time, studies are carried out on inhibition of HMGB1 release in hyperoxia to reduce susceptibility to pneumonia [110]. Also, an increase of HMGB1 concentration in many tumors was observed. It is believed that tumor progression depends on extra-cellular HMGB1 [111]. According to studies, HMGB1 protects tumor cells against apoptosis, suppresses signaling of cell growth control, and exhibits angiogenic activity [112]. At the same time, HMGB1 is a significant protein involved in regeneration of tissues, particularly after chemotherapy and radiotherapy. The total quantity of HMGB1 in serum informs about biomarker during post-traumatic injuries models. HMGB1 is late mediator of sepsis when becomes to dysfunction of organs [110]. Although HMGB1 demonstrates features of prognostic marker, the reduction of its concentration should be the main therapeutic goal, due to its impact on deregulation of proinflammatory and anti-inflammatory response [82].
MicroRNA
A recent researches regarding a sensitive, early biomarker were focused on genetic markers. MicroRNA (miRNA, miR) are short, single-thread fragments of RNA containing 21-23 nucleotides. miRNA are coded in cell genome and transcribes the same mRNA. They are present in protein complex and are involved in endogenous, negative regulation of other gene expression. miRNA are capable of protein translation of specific interference and mRNA degradation intensification. miRNA contribute in maintaining organisms’ homeostasis, in the control of cells physiological functions, cell cycle, differentiation, and apoptosis. Changes of miRNAs concentration were observed during some diseases: sepsis, diabetes, tumors, chronic obstructive pulmonary disease, asthma [43], inflammatory bowel disease, and schizophrenia [113].
The selected microRNA were recognized as potential biomarkers of early sepsis, mainly due to their role during immune response. Additional benefit of microRNA relates to possible determination thereof in serum and plasma samples [114]. Some study included measurement of miRNA in blood of patient with diagnosed sepsis, but the results of the study were inconsistent [115].
MicroRNA control both genes related to pro-inflammatory and anti-inflammatory immune response. Overexpression of miR-486, miR-182 was observed in septic patients comparing to healthy people and during sepsis, the expression reduced followed by miR-150, miR-342-5p. A correlation was shown between miR-150 and results in SOFA scale (sequential organ failure assessment), which confirmed suitability of this marker due its correlation with disease progression, and possible differentiation of sepsis and severe sepsis. Moreover, miR-150 concentration in plasma (determined in relation to miR-192) depended on number of leukocytes. miR-150 controls expression of genes responsible for early immune response. The correlation was indicated regarding miR-150 expression with expression of TNF-α, IL-10, and IL-18 genes. [113]. The conducted research specified five types of miR (miR-30d-5p, miR-30a-5p, miR-192-5p, miR-26a-5p, miR-23a-5p), enabling differentiation of sepsis and SIRS. This research confirmed also negative correlation between miR concentration and concentration of pro-inflammatory cytokines [115]. According to a study including patients’ serum with diagnosed sepsis, miR-16, miR-193b, and miR-483-5b were determined as biomarkers of potential prognostic value; the highest concentration was observed in patients who died [43].
Studies on mice indicated an essential impact of miR-155 and miR-125b on innate immune response. Studies revealed that macrophage stimulation by LPS resulted in increased expression of miR-155 and reduced expression of miR-125b. Obtained results indicated that control of TLR4 signaling pathway, from activation with LPS until production of TNF-α, was dependent on miR-155 and miR-125b, which was promising with regard to establishment of a new therapy [116].
Conclusions
Above results of research on efficient biomarker or set of biomarkers for severe infection indicated a complex and still unknown pathomechanism of sepsis as well as many requirements for marker substances. At present, the most promising are genetic markers (including microRNA) or multimarkers (simultaneous determination of various parameters, e.g. PCT, CD64, and sTREM-1) [117], providing a potential regarding predicting or recognition of severe infection at early phase. Application of such biomarkers would be crucial with respect to monitoring of complications (e.g. post-operation events) or higher risk groups, e.g. in patients after major oncology operations. The essential element of these studies shall be the impact of disease-related malnutrition (demonstrated by as many as 100% patients admitted to ICU) with regard to measured parameters [118].
In patients with diagnosed sepsis, it is likely that the early diagnostics regarding immune disorder and better understanding of immune response deregulation will enable to administer treatment, anticipating injury of some organs resulted inter alia from excessive activation of immune system cells. Unfortunately, a rapid progress in researches regarding an efficient therapy of severe infection is unlikely to happen, due to imperfect sepsis models used during experimental studies. As is known, mice and humans feature with quiet different immune system [119]. There is also a lack of randomized trials regarding regulatory options for early immune response as well as monitoring of sepsis. Severe infections are associated with dynamic progress, coexistence of SIRS and CARS, and heterogeneity of patients’ group. Future studies should take into account maximally uniformed groups and monitoring of immune system dynamic response that has an impact on the course of disease and prognosis.