
Introduction
Neurofibromatosis type 1 (NF1) is a multisystem disorder caused by an inactivating mutation in the gene encoding neurofibromin 1, a protein that functions as a tumor suppressor. In approximately half of the cases, the mutation occurs de novo, while familial cases are inherited in an autosomal dominant manner. The condition is characterized by 100% penetrance but variable expression, both throughout an individual’s life and among affected family members [1, 2].
The diagnosis of neurofibromatosis type 1 is based on the presence of at least two of the seven diagnostic criteria established by the National Institutes of Health (NIH) [3]. Genetic testing for the identification of a pathogenic NF1 gene variant may be helpful.
Clinical criteria include café-au-lait spots, neurofibromas, Lisch nodules in the iris, axillary or inguinal freckling, optic pathway glioma, characteristic bone lesions, and a first-degree relative diagnosed with NF1 [3].
The aim of this study is to highlight the endocrine disorders observed in patients with neurofibromatosis type 1, which – although sometimes underrepresented in the literature and in daily clinical practice – constitute a significant aspect of the disease and must be taken into account in the comprehensive care of this patient group.
Material and methods
A retrospective analysis was conducted on 66 medical records of pediatric patients (up to 18 years of age) hospitalized at the University Hospital between 2018 and 2024 with a diagnosis of neurofibromatosis type 1. The study included patients who met the diagnostic criteria for NF1 according to the NIH guidelines and with pathogenic variant in the NF1 gene identified in a genetic test [3]. Patients with insufficient data for comprehensive analysis (e.g., missing genetic test results or incomplete medical records) were excluded from the study. Clinical data included NF1 manifestations, anthropometric measurements (weight and height), pubertal stage, and available imaging results. Growth was evaluated using age- and sex-specific percentile charts [4], and pubertal development was assessed according to Tanner staging [5]. Endocrine disorders were diagnosed according to standard pediatric clinical and laboratory criteria. Precocious puberty was defined as the onset of pubertal signs before 8 years in girls and 9 years in boys [6]. GnRH-dependent precocious puberty was diagnosed by elevated basal LH (> 0.3 IU/l) predominating over follicle-stimulating hormone (FSH), with increased estradiol in girls and testosterone in boys; confirmation was obtained via a GnRH stimulation test, where a peak LH > 5.0 IU/l predominating over FSH indicated true GnRH-dependent puberty [6]
The data were processed using descriptive statistics, calculating the prevalence of endocrinological disorders within the analyzed patient group.
This study focuses on a detailed description of two pediatric patients selected from this cohort who developed significant endocrine disorders associated with NF1 that required treatment. These cases were chosen to illustrate the clinical course, diagnostic challenges, and consequences of delayed treatment.
Results
Out of the 66 pediatric patients studied, 22 (33.3%) with NF1 were hospitalized in the Department of Pediatrics, Endocrinology, Diabetology, and Metabolic Diseases.
The remaining patients were treated in other departments, the majority of them in the Department of Oncology. Patients were referred to the Department of Pediatrics, Endocrinology, Diabetology, and Metabolic Diseases for the following health issues: short stature (8 patients, 36.36%), precocious puberty (5 patients, 22.72%), rapid progression of puberty (1 patient, 4.5%), primary amenorrhea/low body weight (1 patient, 4.5%), hyponatremia (1 patient, 4.5%), hypernatremia (1 patient, 4.5%), hypothyroidism (1 patient, 4.5%), arterial hypertension (1 patient, 4.5%), obesity (1 patients, 4.5%), and routine hormonal evaluation due to the underlying disease (2 patients, 9.09%).
Endocrine disorders requiring treatment were identified in 6 children (27.27%) from 22 patients hospitalized in the endocrinology unit.
In 5 patients (22.72%), GnRH-dependent precocious puberty was diagnosed, which was associated with the presence of gliomas involving the optic pathway and/or hypothalamus, visible on magnetic resonance imaging. In one patient (4.5%), multiple pituitary hormone deficiency and diabetes insipidus were diagnosed, resulting from surgery of a suprasellar tumor – pilocytic astrocytoma World Health Organization grade I, confirmed by histopathological examination.
Patients with GnRH-dependent precocious puberty were enrolled in a long-acting GnRH agonist treatment program (reimbursed by the National Health Fund in Poland) [7]. These drugs act on pituitary gonadotropin receptors, causing continuous stimulation that leads to receptor desensitization and suppression of the hypothalamic–pituitary–gonadal axis [8]. Clinically, this results in arrest of further pubertal progression, slowing of bone age advancement, and reduction of growth velocity.
Triptorelin was administered intramuscularly every 28 days: children weighing < 20 kg received half of a 3.75 mg SR ampoule, while children ≥ 20 kg received a full ampoule [7]. The mean age at treatment initiation was 7 years. NF1 patients accounted for 4.5% of all children treated with triptorelin during the analyzed period, including those treated for other causes of precocious puberty.
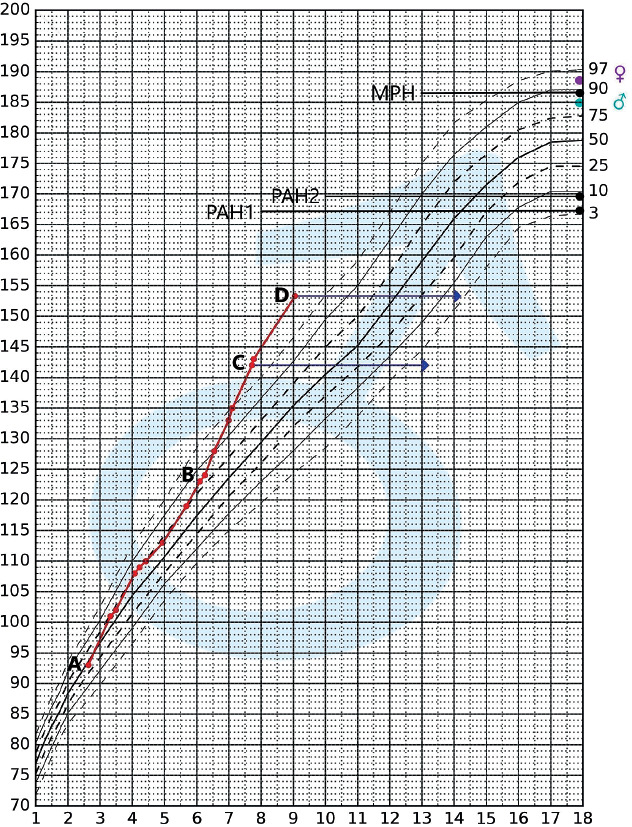
Particular attention in our analysis was drawn to 2 boys referred to the clinic for evaluation of precocious puberty, who presented with advanced pubertal features and significantly accelerated bone age. Neither boy had a family history of NF1 (de novo mutation), and both had café-au-lait spots (CAL) observed since birth. In patient 1, NF1 was diagnosed at the age of 1.5 years, when brain MRI revealed an optic pathway glioma involving the optic nerves and chiasm. Patient 1 was first admitted to the Endocrinology Clinic at the age of 7 years and 10 months (2021). Bone age was assessed at 14 years (standard deviation score [SDS] +6.2) [9], corresponding to an advancement of approximately 6.2 years relative to chronological age. Predicted adult height (PAH) [4] was 167.06 cm, which was 19.44 cm (SDS –2.99) [10] below the mid-parental height (MPH) [4] of 186.5 cm. Testicular volume measured by palpation was 15 ml.
Patient 1 was enrolled in the triptorelin treatment program. Therapy was discontinued once the patient reached a chronological age appropriate for the onset of puberty, with a bone age [9] of 14 years and a PAH [4] of 169.39 cm, representing an improvement of +2.33 cm in growth prognosis compared to the pre-treatment assessment.
The patient’s growth curve is presented in the percentile chart below (Fig. 1).
Figure 1
Percentile chart according to Palczewska and Niedźwiecka [4] with the growth curve of patient 1 plotted
Marked points: A – time of NF1 diagnosis; B – onset of accelerated growth; C – first hospitalization in the Endocrinology Clinic; D – end of triptorelin treatment; → – height in reference to the patient’s bone age
MPH – midparental height (186.5 cm); PAH1 – predicted adult height before initiation of triptorelin therapy (167.1 cm); PAH2 – predicted adultheight after completion of triptorelin therapy (169.4 cm); ♀ – mother’s height; ♂ – father’s height.
Patient 2 was admitted to the clinic at the age of 6 years and 1 month (2023). On the day of admission, NF1 was diagnosed based on NIH clinical criteria [3]. Physical examination revealed numerous café-au-lait spots, cutaneous, subcutaneous, and plexiform neurofibromas (confirmed on MRI). Testicular volume measured by palpation was 12 ml. Bone age was assessed at 12.5 years (SDS +6.4) [9], corresponding to an advancement of approximately 6.4 years relative to chronological age. Predicted adult height [4] was 163.4 cm, which was 15.1 cm (SDS –2.32) [10] below the MPH [4] of 178.5 cm. Brain MRI revealed a hypothalamic glioma involving the optic chiasm and the initial segments of the optic tracts. Patient 2 is currently undergoing triptorelin treatment.
These 2 cases were selected from the cohort to illustrate the clinical course, diagnostic challenges, and consequences of delayed treatment of endocrine disorders in children with NF1.
Discussion
Endocrine disorders represent a significant clinical aspect in children NF1, although they may still be underrecognized in everyday clinical practice. In our retrospective analysis of 66 patients diagnosed with NF1, 27.27% were found to have hormonal disorders requiring treatment. The most common issue in this group was central precocious puberty (GnRH-dependent), associated with optic pathway and/or hypothalamic gliomas (22.72%).
Similar observations were reported in a retrospective study including 181 children with NF1 in Portugal, published in 2025 in the European Journal of Endocrinology. The overall prevalence of endocrine disorders in this cohort was 23.2%, and the most frequent manifestation was precocious puberty (12.2%), which occurred more often in children with optic pathway gliomas (19.2% vs. 7.4%). The study also showed that precocious puberty may occur in NF1 patients without central nervous system (CNS) lesions [11].
In another study including 45 children with NF1, published in 2023 in the journal Hormones (Athens), it was also found that precocious puberty can occur independently of the presence of CNS lesions [12]. The findings of both studies complement our analysis.
Patient 1 from our cohort, whose treatment with triptorelin was initiated only after the age of 7 years (in 2021), despite a diagnosis of NF1 and an optic pathway glioma at 1.5 years of age, illustrates the consequences of delayed endocrine intervention. Therapy was started when the patient already exhibited advanced signs of puberty and markedly accelerated bone age, negatively affecting growth prognosis. The delay in diagnosis and treatment may have been related to the COVID-19 pandemic, which significantly impacted healthcare delivery, particularly for patients with chronic conditions.
Our observations, together with the literature data [11, 12], underscore the need for regular developmental assessment in children with NF1, including monitoring of pubertal progression and growth rate, regardless of the presence of CNS lesions. Recommendations from the American Academy of Sciences [2], the European Reference Network for Rare Genodermatoses and Neurofibromatoses [13], and the Eastern Paediatric Epilepsy Network (part of the Organisation of Paediatric Epilepsy Networks in the UK) [14] advise annual assessment of pubertal development in children with NF1. However, these guidelines do not differentiate patients based on age or the presence or absence of CNS lesions.
Literature data also indicate that optic pathway gliomas occur in approximately 15% of children with NF1 before the age of 6 [15], whereas the development of new gliomas is rare in older children and adults [16, 17]. However, the Optic Pathway Task Force does not recommend routine screening MRI for all patients with NF1 [18].
This raises the question of whether developmental assessments (including pubertal staging and growth monitoring) in younger patients and in those with confirmed lesions involving the optic pathway and/or hypothalamus should be performed at shorter intervals than the standard annual evaluation, for example every 6 months. This mainly applies to girls up to 8 years old and boys up to 9 years old. Such a monitoring schedule could facilitate earlier detection of endocrine disorders and allow timely therapeutic intervention, potentially improving treatment outcomes, final adult height, and quality of life.
The limitations of our study include its retrospective design, occasional incomplete data that hinder the formulation of broader conclusions, and the relatively small number of patients requiring endocrine therapy. Nevertheless, our observations provide meaningful clinical insights that may contribute to improving care for patients with NF1, underscoring the importance of early diagnosis and treatment of pubertal disorders in this population.
Conclusions
One of the most common endocrinological disorders in patients with NF1 is GnRH-dependent precocious puberty, primarily due to gliomas in the optic pathway and/or hypothalamus. Early diagnosis and prompt initiation of treatment with long-acting GnRH agonists are essential to achieve optimal therapeutic outcomes, particularly in terms of final adult height.
Annual assessment of somatic development, including sexual maturation and growth velocity, should be an integral part of comprehensive care for children with NF1. In our opinion, in patients with the described CNS changes, as well as in younger children without imaging, these evaluations should be conducted more frequently – at least every 6 months. This mainly applies to girls up to 8 years old and boys up to 9 years old.
To improve the quality of care for patients with NF1, it is necessary to continue developing integrated diagnostic and therapeutic pathways, which will involve collaboration among specialists from various medical fields.








