

Introduction
Myelodysplastic syndromes (MDS) represent a heterogeneous group of clonal hematological malignancies characterized by the presence of cytopenias resulting from ineffective hematopoiesis, multilineage dysplasia and high risk of transformation to secondary acute myeloid leukemia (secAML) [1, 2]. Whole genome sequencing identified more than 45 genes involved in MDS molecular pathophysiology, which encode proteins involved in pre-mRNA splicing (SRSF2, SF3B1, U2AF1, ZRSR2), DNA methylation (TET2, DNMT3A, IDH1/2), histone modification (ASXL1, EZH2), signal transduction and the TP53 pathway. Among them also cohesion components (SMC1A, RAD21, STAG1, STAG2, SMC3) and transcription factors (RUNX1, ETV6, GATA2) were found [1, 3]. Various somatic mutations were associated with overall prognosis and specific MDS morphological features [2]. Understanding molecular mechanisms of genetic disease evolution may allow for targeted MDS treatment. Measurable residual disease (MRD) and donor chimerism (DC) monitoring remain essential in the management strategy of MDS patients after allogenic hematopoietic cell transplantation (alloHCT). Next generation sequencing (NGS) application enables detection of sequence variants and their further monitoring. Therefore in this study we intended to evaluate the diagnostic utility of targeted NGS, Sanger sequencing (SSeq) and pyrosequencing in monitoring mutational dynamics with samples collected sequentially at different stages of MDS/secAML.
Material and methods
DNA was isolated from bone marrow samples (BM), peripheral blood samples (PB) and saliva samples (SAL) by a standard phenol : chloroform procedure. Samples collected in 2009-2018 from a patient with MDS/secAML were analyzed in order to examine the mutational profile during the course of the disease (Fig. 1). DNA samples isolated from PB of six healthy controls were used to establish the limit of allele frequency (AF) detection by pyrosequencing. Molecular analysis was performed in accordance with the Declaration of Helsinki and Ethics Committee approval (563/14) with written informed consent obtained from the patient. Both MDS and secAML diagnoses were established according to the 2016 World Health Organization criteria [2]. Treatment response and cytogenetic and genetic risk were evaluated according to the 2017 European LeukemiaNet (ELN) recommendations [4].
Fig. 1
A) Data concerning percentage content of bone marrow blasts, ring sideroblasts, peripheral blood chimerism, implemented treatment, performed molecular analysis and additional information during the course of the patient’s disease. B) Allele frequency results for selected DNA sequence variants (obtained by NGS and pyrosequencing) in BM, PB and SAL as well as information about AF for healthy control and AF cut-off value for selected DNA sequence variants. C) Electrophero- grams presenting results of pyrosequencing (c.1945G>T ASXL1; c.1874G>T SF3B1; c.4044+2dupT TET2; c.4076G>T TET2) performed in: I – May 2009, II – January 2017, III – May 2018
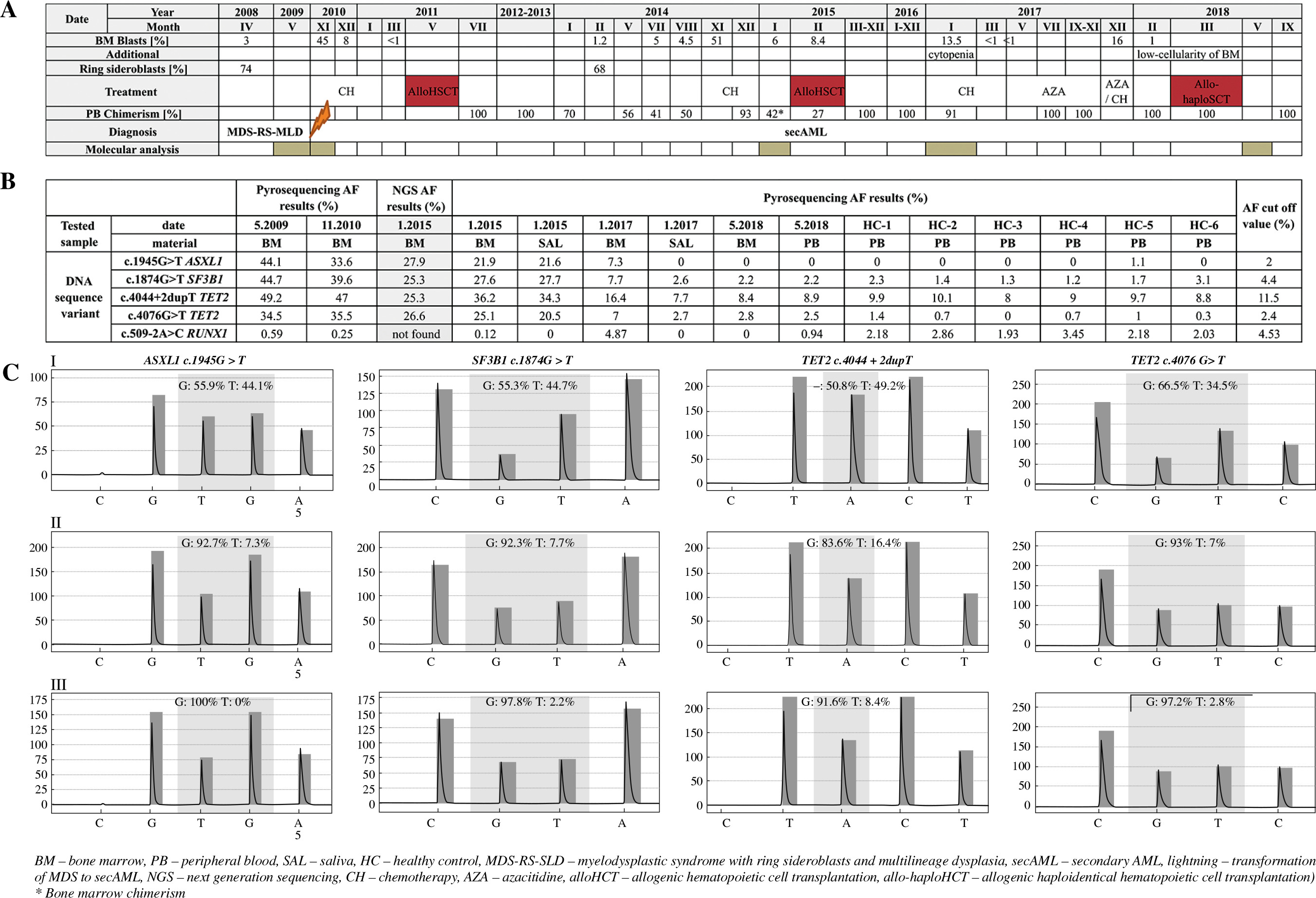
Targeted NGS was performed in a BM sample taken in January 2015 at the stage of secAML relapse after the first alloHCT. 67 exons (ex.) (91 amplicons) of nine genes – SF3B1 (ex. 12-16), U2AF1 (ex. 2,6), SRSF2 (ex. 1), ZRSR2 (whole coding region), TET2 (ex. 3-11), ASXL1 (ex. 12), DNMT3A (ex. 2-23), TP53 (ex. 2-11) and RUNX1 (ex. 3-8) – were paired-end sequenced using the targeted NGS (MiSeq system (Illumina)). The sequencing coverage accounted for 31-9095 with mean 250 read length. Bioinformatic analyses comprised mapping reads using the bwa-mem algorithm (BWA, 0.7.10) [5] to the reference genome UCSC Genome Browser (GRCh37/hg19). The detection of variants was performed using GATK (3.5) software [6] with their further annotation completed by snpEff (4.2) [7] based on the dbSNP142 database. For further validation we selected only nonsynonymous substitutions and indels, novel variants or those already annotated with mean AF < 1%. Next, detected variants were confirmed with SSeq and pyrosequencing. Primer sequences, amplicon lengths and localization and PCR conditions are listed in Table 1. Pyrosequencing was carried out according to the manufacturer’s protocol at PyroMark Q48 Autoprep (Qiagen). SSeq was performed according to the protocol described by Kiwerska et al. [8]. The obtained results were analyzed in relation to the patient’s clinical data.
Table 1
Primer sequences, genomic localization of the amplified sequences and reaction conditions
Case report
The patient, a 54-year-old man, was diagnosed with MDS with ring sideroblasts and multilineage dysplasia (MDS-RS-MLD) in April 2008 (3% BM blasts, 74% BM ring sideroblasts, normal karyotype and intermediate disease risk according to the Revised International Prognostic Scoring System (IPSS-R)). In November 2010 due to transformation to secAML the patient underwent conventional chemotherapy with daunorubicin and cytarabine (without hypomethylating agents) followed by alloHCT (May 2011). Second alloHCT (preceded by treatment with chemotherapy with daunorubicin and cytarabine) was performed in February 2015 and third alloHCT (preceded by hypomethylating agents) was performed in March 2018 (Fig. 1). Targeted NGS was performed in a BM sample taken at the moment of the resistance to the implemented therapy for the secAML relapse (January 2015; with PB DC = 42% and BM blasts = 6%) and revealed the presence of the following DNA sequence variants: c.4044+2dupT (splicing variant) and c.4076G>T (p.Arg1359Leu) in TET2, c.1945G>T (p.Gly649*) in ASXL1 and c.1874G>T (p.Arg625Leu) in SF3B1, with AF of: 25.3%, 26.6%, 27.9%, 25.3%, respectively. Presence of all detected variants was confirmed using SSeq and pyrosequencing in both BM and SAL. For each DNA sequence variant the sensitivity of the pyrosequencing method (AF percentage above which the tested sample is considered as positive) was established. To do so, twice the standard deviation of AF for healthy controls + the highest value of AF observed in controls was counted. The cut-off values for c.4044+2dupT TET2, c.4076G>T TET2, c.1945G>T ASXL1 and c.1874G>T SF3B1 DNA variants were determined as: 11.5%, 2.4%, 2%, 4.4%, respectively. AF values obtained by pyrosequencing corresponded to those obtained by NGS. Moreover, for analyzed DNA sequence variants, AF levels obtained by pyrosequencing in BM and SAL were very similar (Fig. 1).
In retrospective analysis of BM (May 2009; November 2010) with pyrosequencing and SSeq, c.4044+2dupT and c.4076G>T TET2, c.1945G>T ASXL1 and c.1874G>T SF3B1 DNA sequence variants were detected. In May 2009 at the MDS stage of disease, AF was 49.2%, 34.5%, 44.1%, 44.7%, respectively (Fig. 1). All detected variants were reported in hematological malignancies according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database [9]; however, except for c.1874G>T SF3B1, the remaining three variants had not been identified in MDS patients so far. In November 2010 transformation to secAML occurred; the patient underwent chemotherapy and was qualified for alloHCT. Using pyrosequencing, AF of above-mentioned variants was determined as 47%, 35.5%, 33.6% and 39.6%, respectively (Fig. 1). AlloHCT from an unrelated donor was performed in May 2011 in complete remission (CR) according ELN 2017 treatment response criteria. Since January 2014 loss of PB DC (70%, 56%, 41%) has been observed and the patient was considered for second alloHCT, but relapse of the disease was diagnosed with 51% of BM blasts. The patient received chemotherapy, but he did not achieve CR according to the ELN 2017 treatment response criteria (no CR). Second alloHCT from the same donor was performed in February 2015 with 8.4% BM blasts and PB DC 27%. Two years later (January 2017) pancytopenia, 13.5% of BM blasts and PB DC of 91% were reported. In BM and SAL obtained from the patient in January 2017 none of the previously detected DNA sequence variants were found with SSeq due to the high DC. On the other hand, all analyzed variants were confirmed in BM with pyrosequencing. In SAL DNA sequence variants were undetectable, probably due to concurrent high DC at disease relapse. Based on leukemic evolution theory and the possibility of additional mutation acquisition we decided to check whether any additional mutation occurred in a BM sample after second relapse of the disease. Using the pyrosequencing method, the presence of RUNX1 (c.509-2A>C), SRSF2 (c.284C>A), TET2 (c.4638G>C) and DNMT3A (c.1014+1G>T; c.2390A>G) DNA sequence variants was demonstrated. Pyrosequencing revealed the presence of the RUNX1 (c.509-2A>C) DNA sequence variant with AF 4.87%. Importantly, the RUNX1 variant was not observed at earlier disease stages. Salvage chemotherapy was implemented, resulting in CR (according ELN 2017 treatment response criteria) and the patient was qualified for maintenance treatment with azacitidine. Subsequent relapse with 16% of BM blasts occurred; thus the patient received chemotherapy, resulting in CR (according ELN 2017 treatment response criteria). The patient was qualified for allogenic haploidentical hematopoietic cell transplantation (allo-haploHCT) from his daughter (March 2018). Since May 2018 acute graft-versus-host disease with pancytopenia has been observed. BM biopsy revealed low cellularity and DC of 100%. In BM and PB obtained from the patient in May 2018 none of the previously detected variants were found with SSeq and pyrosequencing. Four months after allo-haploHCT the patient died from severe graft-versus-host disease with secondary cytopenia, refractory to the treatment.
Discussion
Genetic findings mentioned above indicated that four sequence variants identified with NGS were detected in patient samples since the MDS phase of disease (May 2009) and were persistently reported after transformation to secAML (November 2010, January 2015, January 2017) with the additional RUNX1 c.509-2A>C DNA sequence variant reported since January 2017. Importantly, c.4044+2dupT, c.4076G>T TET2, and c.1945G>T ASXL1 DNA sequence variants were detected for the first time in MDS. Moreover, the presence of c.4044+2dupT and c.4076G>T TET2, c.1945G>T ASXL1 and c.1874G>T SF3B1 DNA sequence variants was confirmed with pyrosequencing and SSeq in both BM and SAL (January 2015). It should be noted that AF detected during pyrosequencing of BM and SAL was very similar (Fig. 1), which highlights the diagnostic utility of SAL testing. However, it is worth noting that the analysis of the mutation presence in SAL is limited by the low amount of leukemic cells as well as presence of high DC in recipients of alloHCT. With clinical suspicion of disease relapse, BM evaluation remains necessary, but our results suggest the clinical usefulness of noninvasive testing with SAL or PB in monitoring the course of disease. Nevertheless, this should be tested on a larger group of patients first.
Comparing pyrosequencing AF results obtained in May 2009, November 2010 and January 2015 and taking into account DC loss (2015 – 42%), we observed a slight AF decrease in tested samples. At the moment of secAML relapse (January 2017) with DC = 91% using the same method, four variants previously identified with targeted NGS, as well as the RUNX1 variant, were detected only in BM but not SAL. Progressive disease in both MDS and secAML is characterized by the emergence of subclones influenced by selection pressure after implemented chemotherapy [10]. In May 2018, after allo-haploHCT (CR, DC = 100%) pyrosequencing results were negative for both BM and PB. Thus, pyrosequencing results corresponded to DC results and additionally introduced information concerning presence of DNA sequence variants in patients after alloHCT. With this approach we are able to confirm the presence of key sequence variants previously detected by NGS. Moreover, the detected DNA sequence variants in selected genes and their prognostic value could influence the therapeutic strategy.
Myelodysplastic syndromes and secAML are very heterogenous diseases and the presence of multiple genetic founding clones translates to different sensitivity to the applied therapy. Moreover, the increasing number of early driver somatic mutations steadily reduces leukemia-free survival [11]. Sequence variants of TET2 and ASXL1 genes are associated with clonal hematopoiesis of indeterminate potential (CHIP). However, CHIP is associated with AF < 10% and presence of single mutations, whereas MDS and AML are associated with higher AF and ≥ 2 mutations [12]. The prognostic significance of TET2 DNA sequence variants in MDS remains unclear [13]. According to Smith et al., TET2 sequence variants in MDS patients do not affect the survival and risk of AML transformation [14]. Furthermore, low expression of the TET2 gene is associated with poor prognosis in MDS patients. Moreover, metaanalysis also revealed that the presence of TET2 DNA sequence variants does not have a significant impact on overall survival. MDS patients with TET2 mutations have increased response rates to hypomethylating agents compared to patients without those mutations. However, treatment with hypomethylating agents does not prolong the overall survival (OS) in MDS patients with TET2 mutations compared to those without. Thus, TET2 status may be used as an additional factor for predicting patients’ response to hypomethylating agents [15]. The SF3B1 variant reported here was found to correlate with ring sideroblasts MDS subtype 2, and this was observed in our case too. Talati et al. associated this variant with favorable prognosis in MDS [16].
Furthermore, patients with SF3B1 mutations showed better treatment responses to erythropoiesis-stimulating agents with no differences in response to hypomethylating agents or lenalidomide [17]. It was also demonstrated that the presence of ASXL1 somatic variants translates to adverse outcome as well as disease relapse and shorter survival after alloHCT in both MDS and MDS/AML patients [18]. It is worth noting that the implemented therapy may also affect time to relapse. The use of anti-cancer treatment results in multiple dynamic changes leading to the subclonal architecture of cancer, driving disease progression and treatment resistance, and is called ‘clonal evolution’ [19]. Mutations in the RUNX1 gene are associated with adverse prognosis in MDS [20], so acquisition of the RUNX1 mutation and its co-occurrence with previously detected DNA variants could reflect disease progression and its influence on the response to the therapy. Importantly, based on Ping Wu’s results, co-occurrence of ASXL1 and RUNX1 DNA sequence variants is related to poor response to hypomethylating agents such as azacytidine or decitabine and their influence on shorter survival [21]. Although the rate of relapse or treatment related mortality is high, alloHCT represents the only curative treatment for high-risk MDS, and it remains crucial to carefully monitor the persistence of genetic alterations after alloHCT [22]. Targeted sequencing of 40 genes in 87 MDS patients undergoing alloHCT identified TET2, DNMT3A and TP53 mutations to be associated with reduced overall survival [23]. Both literature data and our results suggest a significant need to screen MDS patients for the presence of DNA sequence variants in genes of proven clinical relevance. This information could help in treatment management and further course of disease monitoring.
Conclusions
The appropriate MRD monitoring in MDS remains a challenge as the quantity of disease clones is lower than in AML [24]. Our results revealed that the variants previously described for AML (c.4044+2dupT TET2, c.1945G>T ASXL1) were present at the MDS stage of disease. It confirms that the molecular landscape of secAML changes with the disease progression (acquiring c.509-2A>C RUNX1 variant). We suggest: 1) NGS to detect the mutational profile of early MDS and 2) tracking changes in the molecular landscape of MDS/secAML during the course of the disease as essential for improving further treatment. Our paper is the first report of c.4044+2dupT, c.4076G>T TET2 and c.1945G>T ASXL1 DNA sequence variants in MDS.