

Introduction
Rheumatoid arthritis (RA) is a common autoimmune and chronic inflammatory disease which is characterized by chronic inflammation, hyperplasia of synovial tissues, as well as cartilage and bone structural damage [1]. In recent years, many studies have been undertaken to explore the pathogenesis of RA and showed that maturation and migration of dendritic cells (DCs) take a central role in RA pathogenesis.
DCs are powerful antigen-presenting cells (APCs), which take up and process the antigens (Ags), and assemble antigenic peptides in MHC molecules. Along with their maturation, DCs migrate to draining lymph nodes (LNs), where they interact with naive T lymphocytes and shape them to be antigen-specific T cells [2, 3]. DCs also regulate the production of pro-inflammatory mediators and autoantibodies, thereby orchestrating the development of chronic inflammation [4-6]. Therefore, the maturation and migration of DCs are important in the initiation and regulation of the immune responses in RA.
The application of anti-tumor necrosis factor (anti-TNF) biological agents, such as etanercept (ETN), has dramatically improved the outcome of RA patients, including reduced disease activity and functional disability, and decreased joint damage [7]. However, there are scarce data investigating the mechanisms of DCs during the treatment of ETN in RA.
In this study, we investigated the effects of ETN on maturation and migration of DCs in vitro. Furthermore, we assessed the in-vivo effect of ETN on the number of DCs, T and B lymphocytes in LNs of the collagen-induced arthritis (CIA) mouse model, to explore the mechanisms of ETN.
Material and methods
Cell preparation
BMDCs were generated from bone marrow as described [8]. Briefly, bone marrow cells (2 × 106) were flushed out with RPMI 1640 medium from femurs and tibias of male C57BL/6 mice. Cells were cultured for 8 days at 37°C in 5% CO2 in 100-mm-diameter petri dishes (Falcon, Becton Dickinson, Germany) in complete medium, containing 10% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, NY) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, 2 mM L-glutamine, 50 mM 2-ME (all from Sigma-Aldrich) and 20 ng/ml GM-CSF (PeproTech, Rocky Hill, NJ). Then the cells were harvested on day 8. The BMDCs were incubated in culture medium containing 1 μg/ml LPS (Sigma-Aldrich) for 24hrs to induce maturation.
Determination of DC migration in vitro
The effect of ETN on DC migration was determined using a 24-well transwell chemotaxis assay (5-mm pore size, Corning, NY). Briefly, on day 8, BMDCs were collected and treated with 400 U/ml GM-CSF. And then the cells were treated with 10 μg/ml ETN (ETN group), 1 μg/ml Lipopolysaccharide (LPS, named the LPS group), 1 μg/ml LPS +10 μg/ml ETN (ETN added 30 min before LPS, LPS+ETN group), respectively. The cells were collected and plated in the top chamber (2 × 105 BMDCs per well) in RPMI 1640 medium containing 20 mM HEPES (Sigma-Aldrich) 24 hrs later. In the bottom chambers of the transwell plates, 200 ng/ml CCL21 (PeproTech) in RPMI 1640 medium was added. After incubation at 37°C for 3 hrs, DCs migrated to the bottom chambers were harvested and counted by flow cytometry.
Mice
C57BL/6 mice aged 8- to 12-week and Male 6- to 8-week-old DBA/1J mice were housed and bred in the specific pathogen-free (SPF) conditions in the laboratory animal center. The animal experiments were conducted according to the ethical guidelines for animal experiments and under experimental license (No. L2016145).
Induction of collagen-induced arthritis and ETN administration
Collagen-induced arthritis (CIA) was induced as described previously [9]. The bovine type II collagen (CII) solution was dissolved in 0.05 M acetic acid to 2 mg/ml (Chondrex, Inc., USA) and emulsified with an equal volume of Freund’s complete adjuvant (CFA) containing 1 mg/ml Mycobacterium tuberculosis (Chondrex, Inc., USA). On day 0, the DBA/1J mice were subcutaneously immunized with 0.1 ml emulsion at the base of the tail. The booster injection (0.1 ml CII emulsified with Freund’s incomplete adjuvant) was administered near the primary injection site on day 21.
Mice were divided into the control group, CIA group (arthritic mice injected with saline), and ETN + CIA group (CIA mice intraperitoneally injected with 5 mg/kg ETN every 3 days from day 21 to 45).
Clinical assessment of CIA
Mice were subsequently monitored every day for signs of arthritis from the day of the booster injection until the day of sacrifice on day 45. The severity of the joint inflammation was evaluated by clinical scores as follows: 0 = normal, 1 = mild swelling, 2 = moderate swelling, 3 = all joints swelling, 4 = joint distortion and/or rigidity and dysfunction [10].
The clinical scores were the cumulative value for four paws of each mouse, so the maximum score of each mouse was 16. The mice with scores remained stable or enhanced by at least one point for two consecutive days were considered to have arthritis. Two independent observers scored the signs of arthritis.
Immunohistochemistry of DC
Immunostaining of DC in the joint of the mouse was performed with peroxidase labeling techniques. On day 45 the mice were euthanized to obtain hind limbs, fixed in formalin (10%), and decalcified by 5% formic acid. Then the samples were embedded in paraffin, cut to 5-μm slices. Next, the tissue sections were deparaffinized, washed with PBS, and then incubated with the primary Ab against mouse CD11c (Abcam, Cambridge, UK) at 4°C overnight. After washing in PBS, the sections were incubated with HRP-conjugated secondary Ab (DAKO) for 60 min. The sections were stained by immersing in a solution of 0.05% 3,3-diaminobenzidine (Sigma-Aldrich) and 0.01% hydrogen peroxide in 0.05 M Tris (pH 7.4) for 3 min and counterstained with Mayer’s hematoxylin (DAKO) for 2 min. Positive staining was indicated by brownish deposits in a purple background.
Analysis of DC expression and subsets in LNs
Maturation
In the study of DC maturation in vitro, BMDCs were harvested on day 8, and divided into four groups (control, ETN, LPS, LPS + ETN as described above). BMDCs were pre-treated with ETN (10μg/ml) 30 minutes before stimulation with 1 μg/ml LPS, and cultured for 24 hrs. Then the cells were stained with anti-CD11c, anti-CD40, anti-CD80, anti-CD86 and anti-I-Aq (all from BD Pharmingen). The expression of CD40, CD80, CD86 and I-Aq was determined by flow cytometry. In in-vivo study, mice were euthanized on day 45 after primary immunization and LNs harvested for preparation of single-cell suspensions. Cells were stained with monoclonal antibodies for 30 min, and analyzed by flow cytometry.
DC subsets
For determination of DC subsets such as plasmacytoid DCs (pDCs, B220+PDCA+DCs) and myeloid DCs (mDCs, B220-CD11b+DCs), cells from LN were stained with anti-CD11c (BD Pharmingen), anti-B220, anti-PDCA-1 and anti-CD11b (all from BioLegend, CA), then analyzed with flow cytometry.
Assessment of chemokine receptors on blood DCs
Mouse peripheral blood mononuclear cells were harvested and stained with anti-CD11c in association with anti-CXCR4, anti-CCR7 or anti-CCR5 for analysis of the expression of respective chemokine receptors with flow cytometry.
Analysis of lymphocytes of LNs
The LN cells were stained with anti-CD11c, anti-CD3 or anti-CD19 (BD Pharmingen, CA) and analyzed with flow cytometry. The absolute cell numbers of DCs, T cells or B cells were counted by multiplying the total cell number by the respective percentage of DCs, T cells or B cells.
Statistical analysis
The mean arthritis index was analyzed using the nonparametric Mann-Whitney U test. The rest of the data were evaluated using one-way ANOVA. All data were analyzed using SPSS software (version 16.0) and presented as mean±SEM. The p values < 0.05 were considered significant differences between groups.
Results
ETN efficiently inhibited maturation and migration of LPS-treated BMDCs in vitro
The toxicity test showed that ETN at a concentration of 50 μg/ml induced significant BMDC apoptosis and necrosis while ETN at 2 μg/ml or 10 μg/ml did not (Figs. 1A and B). So we used 10 μg/ml ETN in the in vitro experiment.
Fig. 1
ETN efficiently inhibited maturation and migration of LPS-treated BMDCs in vitro. A, B) Bone marrow-derived DCs (BMDCs) were treated with various concentrations of ETN (2 μg/ml, 10 μg/ml, 50 μg/ml) for 24 hrs, and then stained with Annexin V-FITC/Propidium Iodine Apoptosis Detection Kit (Invitrogen, Carlsbad, CA, USA). Apoptotic and necrotic rates were determined by flow cytometry. C, D) BMDCs were treated with LPS (1 μg/ml), ETN (10 μg/ml) for 24 hrs. The expression of CD40, CD80, CD86 and MHC II (I-Aq) were determined by flow cytometry. E) BMDCs were treated with LPS (1 μg/ml) in the presence or absence of ETN (10 μg/ml) for 24 hrs. The cells were collected and plated in the top chamber, 200 ng/ml CCL21 was added in the bottom chambers of the transwell plates. The migrated DCs in the bottom chambers were harvested and counted by flow cytometry 3 hrs later. All data shown were gated on CD11c+cells. * p < 0.05, ** p < 0.01 were for the comparisons between the LPS-treated and untreated DCs, # p < 0.05, ## p < 0.01 were for the comparisons between the ETN-treated and untreated LPS-stimulated DCs
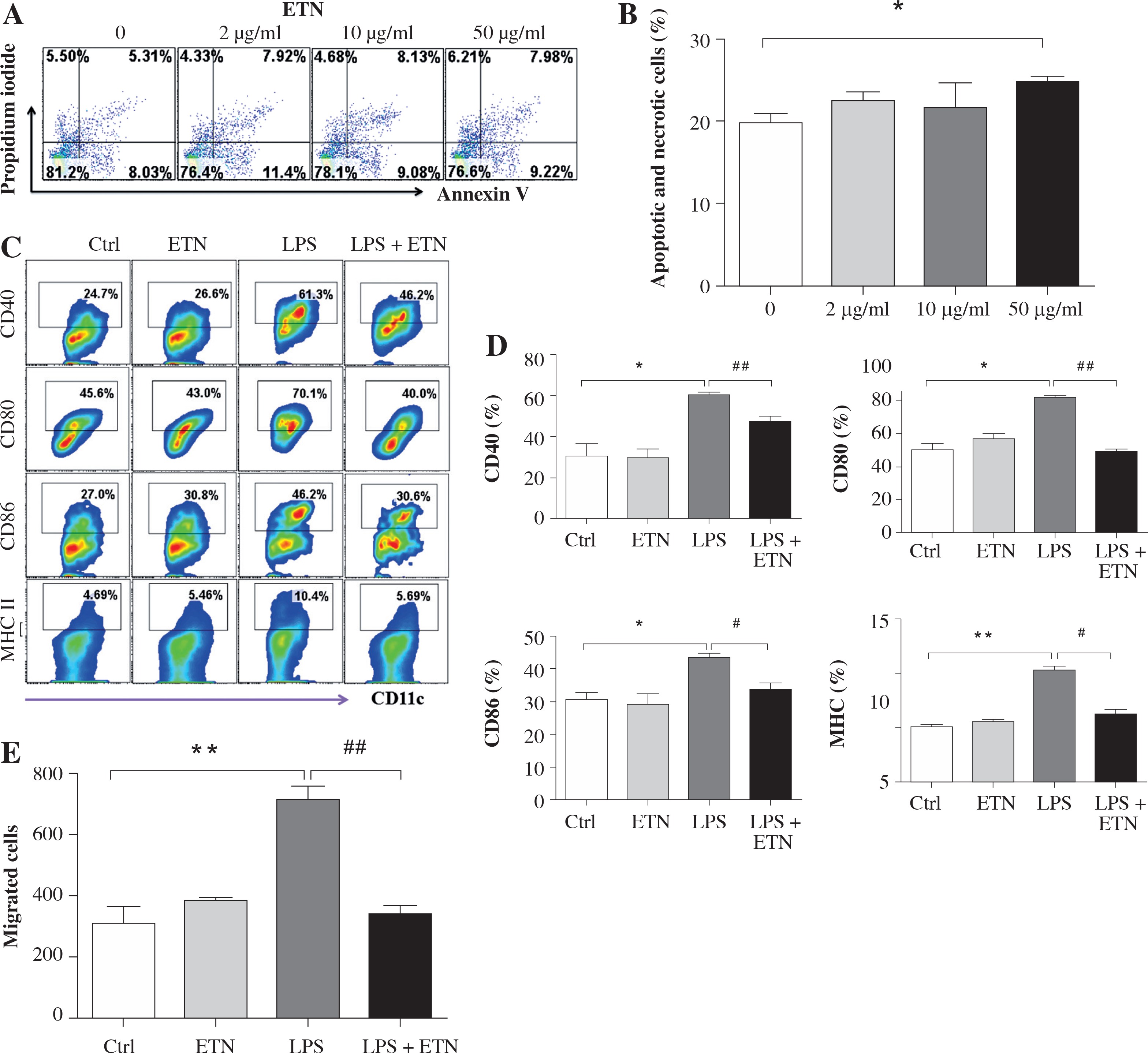
BMDCs were treated with LPS (1μg/ml) to induce maturation, and on day 8 the co-stimulatory molecules CD40, CD80 and CD86 and MHC II (I-Aq) were up regulated dramatically. However, ETN could inhibit the expression of CD40, CD80, CD86 and I-Aq on the CD11c+ cells (Figs. 1C and 1D). Furthermore, the results showed that ETN efficiently decreased the migration of BMDCs attracted by CCL21 (Fig. 1E). These results demonstrated that ETN inhibited BMDCs maturation and migration in vitro.
ETN efficiently delayed the onset and reduced the incidence and the scores of joint arthritis in CIA mice
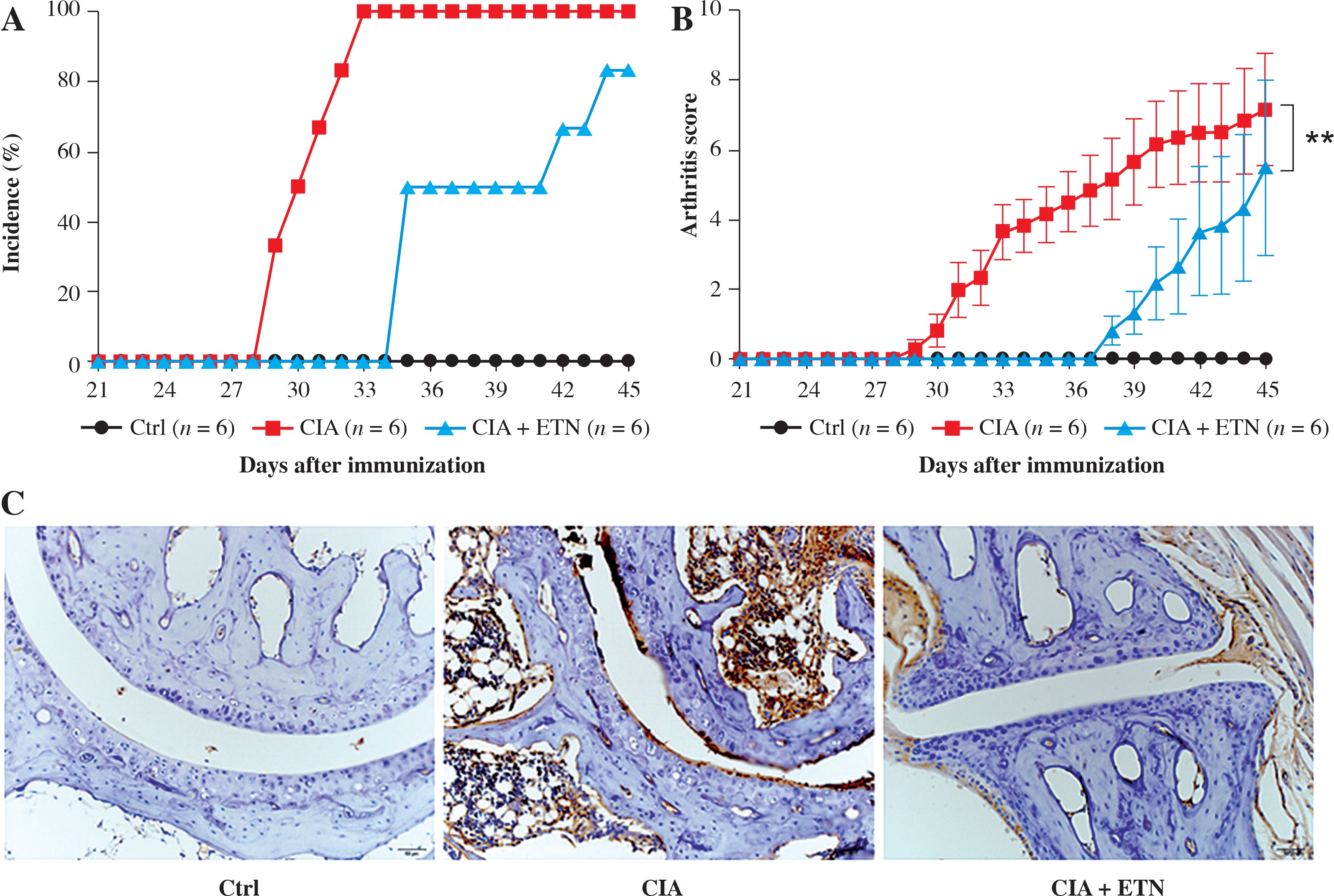
DCs play important roles in the pathogenesis of RA. It is evident that ETN treatment has dramatically improved outcomes in patients with RA through neutralizing TNF-α. However, some studies suggest that the pharmacology of TNF-α blockade is not only anti-inflammatory, but probably more complex than previously thought [11]. To analyze whether ETN regulates the functions of DCs in vivo, we first determined the effect of ETN on an animal CIA model. The results showed that ETN delayed the onset time as well as decreased the incidence of arthritis in the CIA model (Fig. 2A). Moreover, ETN reduced the arthritis scores, indicating that ETN attenuated the severity of CIA (Fig. 2B). Histochemical staining of paws’ joints showed that there were few infiltrating DCs in the joints after ETN treatment (Fig. 2C).
Fig. 2
ETN efficiently delayed the onset and reduced the incidence and scores of joint arthritis in CIA mice. A) ETN significantly decreased the disease incidence as compared with saline-treated CIA mice. B) ETN attenuated the arthritis scores in the CIA model. C) Immunostaining of CD11c of DC in joints (original magnification ×200). ** p < 0.01 was for comparisons between the CIA + ETN treatment group and the CIA model group (n = 6)
ETN treatment inhibited DC maturation and migration in CIA mice
We also evaluated the effect on the maturation of DCs in the CIA model. Our data showed that ETN significantly inhibited the expression of CD40, CD80, CD86 and MHC II (I-Aq) on the surfaces of the CD11c+ cells as compared with the expression in the CIA model group, indicating that ETN suppressed the maturation of DCs in LNs (Fig. 3A).
Fig. 3
ETN treatment inhibited DC maturation and migration in CIA mice. Single-cell suspensions were prepared from the mouse LNs of three groups. A) The changes of mean fluorescence intensity of CD40, CD80, CD86 and MHC II (I-Aq) were analyzed. B) ETN treatment reduced the absolute number of DCs in LNs. C-E) Effects of ETN treatment on CXCR4, CCR5 and CCR7 expression on DCs of peripheral blood in mice. NS, p > 0.05; *p < 0.05, ** p < 0.01 was for the comparison between the healthy control mice and the saline-treated CIA mice, # p < 0.05 was for the comparison between the CIA + ETN group and CIA mice (n = 6)
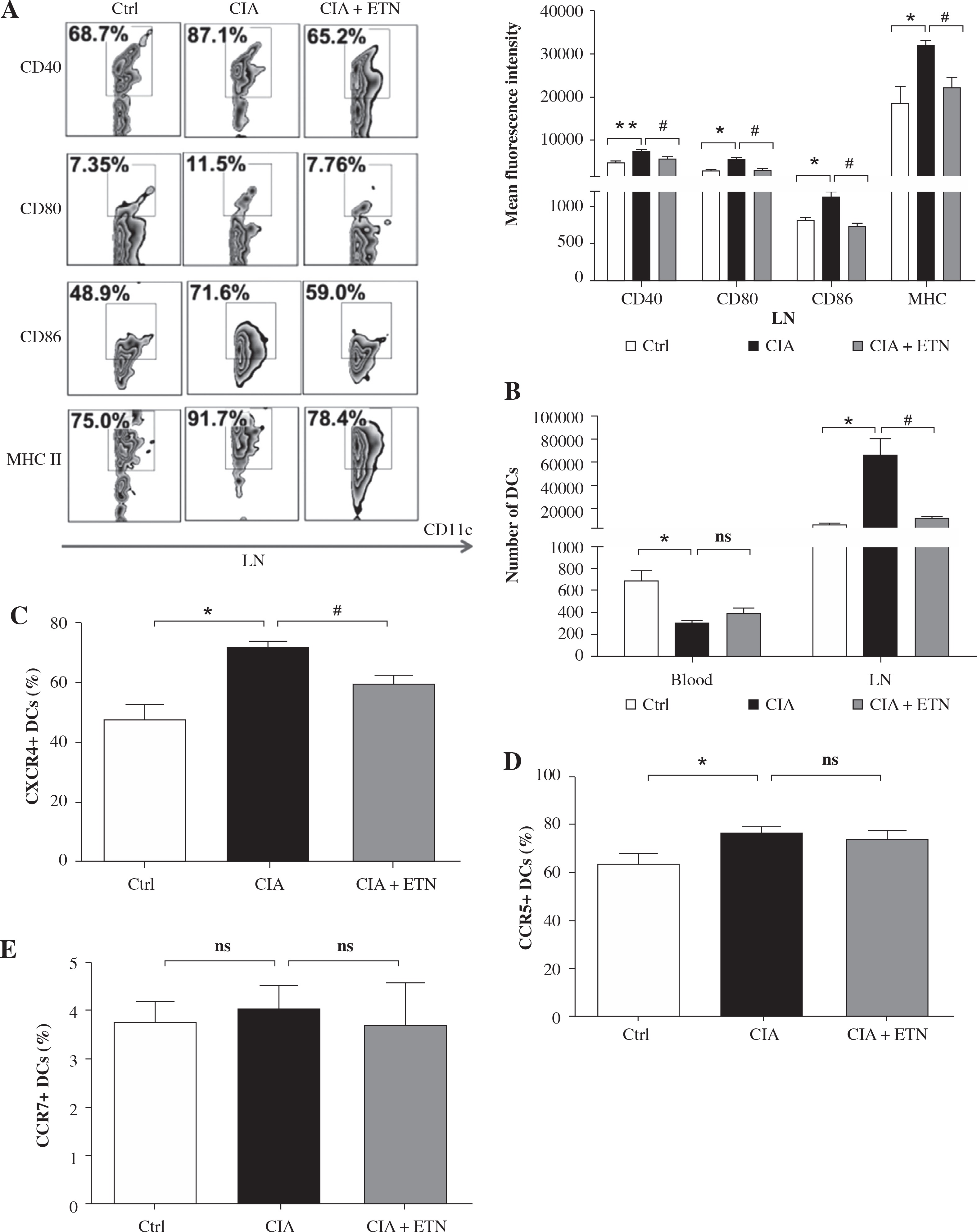
Our results also indicated that the absolute number of DCs in LNs from ETN-treated CIA mice was dramatically reduced as compared with the CIA model group, whereas the number of peripheral blood DCs has no significant differences between the CIA and ETN-treated CIA groups (Fig. 3B). The data indicated that ETN may inhibit DC migration from peripheral tissues to LNs.
The effect of ETN on chemokine receptors of DCs in peripheral blood
Previous studies indicate that some key chemokines and their receptors such as CCR7, CXCR4 and CCR5 are crucial for DC migration to local LNs [12]. We found that ETN reduced the expression of CXCR4 in mouse peripheral blood DCs (Fig. 3C) but had no effect on the expression of CCR5 and CCR7 (Figs. 3D and 3E). Thus, reduced expression of CXCR4 may contribute to the influence of ETN on DC migration.
ETN changed DC subsets of LNs in the CIA model
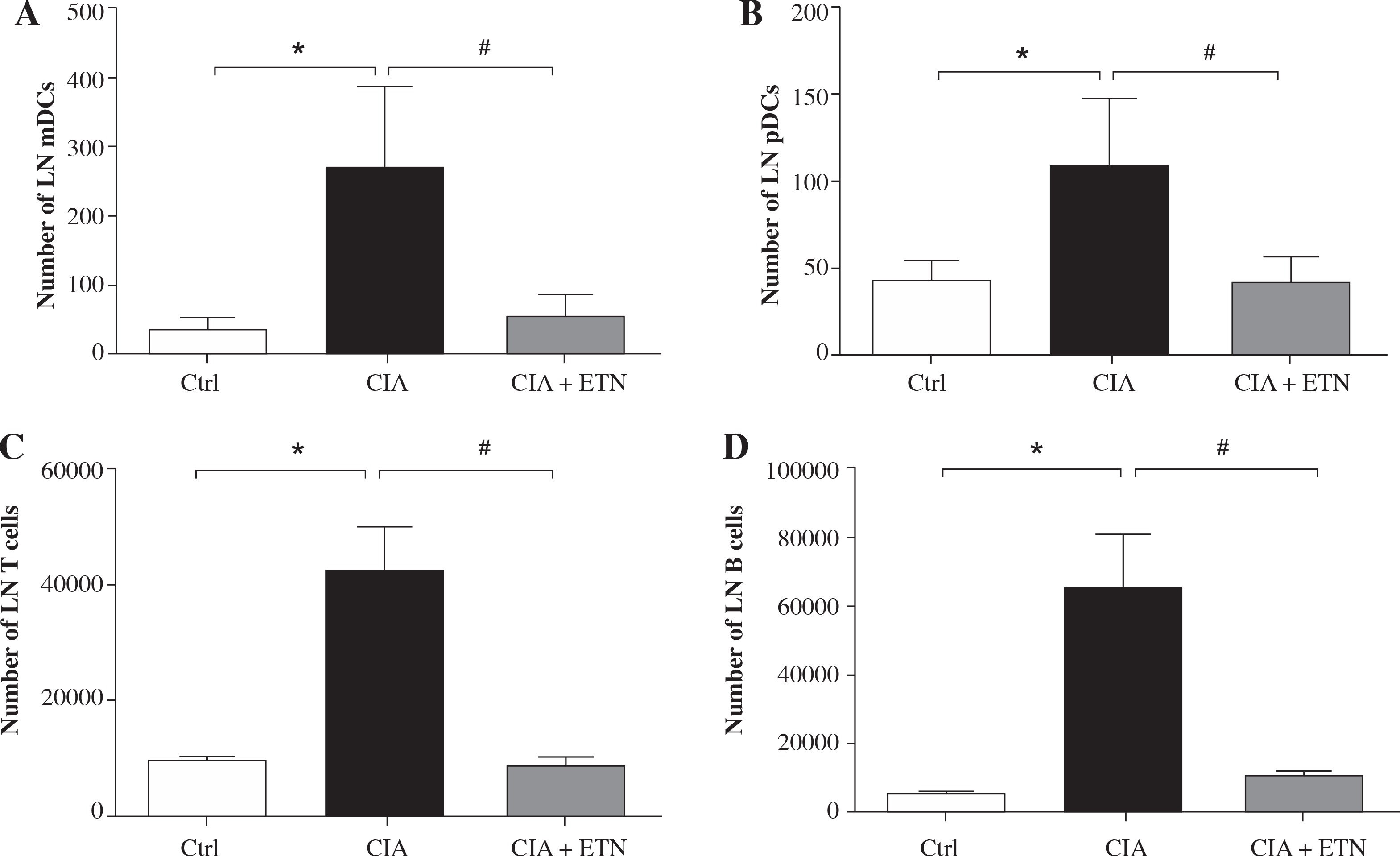
Then we investigated the routes by which ETN affects the migration of DCs to local LNs. As we know, DCs migrate to the draining LNs by different pathways, mDCs traffic through afferent lymphatic vessels while pDCs cross high endothelial venules (HEV) [13]. Our results indicated ETN significantly decreased the number of both mDCs and pDCs in the LNs as compared with that in the CIA group (Figs. 4A and 4B). These results indicate that ETN may change DC subsets in LNs as a result of reduced traffic of DCs to draining LNs by blocking both afferent lymphatic vessel and blood vascular HEV pathways.
Fig. 4
ETN changed DC subsets and cellular composition of LNs in the CIA model. A, B) ETN treatment significantly reduced the number of mDCs (B220-CD11b+) and pDCs (B220+PDCA+) in the LNs. C, D) The absolute number of T cells (CD3+ CD11c+) and B cells (CD19+ CD11c+) in LNs of mice were analyzed by flow cytometry. * p < 0.05 was for the comparison between the healthy control mice and the saline-treated CIA mice, # p < 0.05 was for the comparison between the CIA + ETN group and CIA mice (n = 6)
The effect of ETN on T and B cells in LNs of CIA mice
DCs migrated to local LNs may affect the cell composition and numbers in LNs. We found that ETN could significantly decrease the absolute number of T and B lymphocytes in the draining LNs (Figs. 4C and 4D).
Discussion
RA is an autoimmune and inflammatory disease manifested by the infiltration of DCs, neutrophils, monocytes, T and B lymphocytes and NK cells in synovium [14]. In recent years, many studies have shown that DCs play a central role in the induction of immunity during the pathogenesis of RA [15-17]. Thus, modulation of the functions of DCs, especially DC maturation and migration, remains a potential therapeutic approach to prevent and treat RA [18].
TNF-α is a key player in the pathogenesis of RA and ETN has been demonstrated to have significant efficacy in the treatment of RA by binding TNF and blocking its downstream signal transduction [19]. Some studies have investigated in vivo effects of ETN on various cell types, including the effect on DCs. In recent decades, studies have demonstrated that TNF inhibition is effective in clearing skin lesions by reducing the number of DCs or inducing apoptosis of dermal DCs in psoriasis patients [20, 21]. A study demonstrated that in RA patients, ETN treatment affected the lymphoid germinal centers and follicular DC networks, which contributed to the suppression of memory B lymphocytes [22]. Another study showed that ETN therapy blockaded the maturation of mDCs, increased the number of regulatory T cells, and inhibited the functions of effector T cells [23]. ETN reduced the reactivity of T cells and enhanced DC number in peripheral blood in ankylosing spondylitis patients [24]. Data from Baldwin et al. research showed that TNF-α blockade had effects on survival and function of DC in RA [23], which is consistent with our findings. Moreover, animal experiments also showed that the inhibition of DC function abrogated collagen-induced arthritis [25, 26]. However, all the studies have made little effort about the effect of ETN on DC migration. As we known, DCs migrate to draining LNs following Ag uptake, interact with naive T cells and shape them to be antigen-specific T cells, and then trigger the immune response [27, 28]. Therefore, our study explored the effects of DC on both maturation and migration. The data showed that modulated migration of DC to local LNs may be the effect of ETN on the CIA mice model.
Our results showed that ETN efficiently inhibited the maturation of DC both in vitro and in vivo. ETN treatment delayed the onset, reduced the severity of arthritis and infiltrating DCs in the joints in CIA mice. Moreover, ETN treatment strongly down regulated the number of both mDCs and pDCs in LNs. The down-regulation of CXCR4 might explain at least partly the reason for ETN to block DC migration to draining LNs. Further, ETN dramatically reduced the number of T cells and B cells in LNs as compared with the CIA model group, indicating that ETN may affect T cell/B cell differentiation or proliferation.
mDCs and pDCs are two subsets of DCs, and previous researches have demonstrated that DC migrate into LNs through lymph circulation and blood circulation. mDCs traffic into the LNs by afferent lymph flow, while pDCs migrate to the LNs through high endothelial venules (HEVs) [29, 30]. mDCs are the main APC to stimulate lymphocytes and initiate specific immune responses [27], while pDCs specialized in releasing cytokines and chemokines contribute to the modulation of the strength and duration of T cell, B cell and NK cell responses. Therefore, pDCs play an important role in perpetuation of diseases [31]. To determine the effects of ETN on DC migration in RA, we analyzed the number of mDCs and pDCs in the LNs of ETN- or saline-treated CIA mice. We found that in the CIA model, ETN treatment significantly reduced the number of both mDCs and pDCs of the LNs, indicating that ETN may inhibit the traffic of DCs to the LNs through both the afferent lymph vessels and blood vascular HEVs. Therefore, ETN affects not only the secretion of inflammatory cytokines but also the capacity of antigen presentation.
In recent years, the tolerogenic DCs expressing indoleamine 2,3-dioxygenase1 (IDO1) have already been reported to promote tolerance in some studies [32, 33]. Our study focused on the function of inflammatory DCs, which produce large amounts of cytokines such as IL-12 and IL-23, in inflammatory infiltrates of RA synovial tissue. Our data showed that ETN inhibited the functions of inflammatory DCs and abrogated CIA, but whether ETN could inhibit the function of tolerogenic DCs is not known.
However, the TNFi therapy in RA was associated with a dose-dependent increased risk of serious infections and malignancies [34, 35], thus exploring the mechanisms of TNF blockade is important. Down regulation of antigen presentation by blocking DC maturation and migration capacity might explain at least partly the reason for reduced resistance to infection in TNFi-treated patients. Thus, more researches for a better understanding of TNF-i at the various steps of immune responses may be highly significant for the application of TNFi.