

Introduction
In this article, in the light of the latest findings, we describe the cross-talk between mast cells (MCs) and pathogenic microorganisms.
MCs are versatile effector cells which participate in the immune response and can perform important roles in host defense versus certain parasites (helminths) and in natural immunity to bacterial infection associated with Th2 and IgE. They can phagocytose and kill bacteria (even if in a different manner than macrophages), process other protein antigens, and can function as antigen-presenting cells (APCs). Once activated, MCs perform pro-inflammatory action with cytokine and chemokine generation. By performing this phagocytic function, MCs can release inflammatory cytokines that damage tissues and favor the infectious process. Among inflammatory cytokines they can generate a diverse array of mediators including tumor necrosis factor (TNF) and interleukin (IL)-1 family members which, if inhibited, can also improve the infectious state.
Mast cells
MCs are ubiquitous in the body and are not known to develop allergic reactions or inflammation [1].
MCs are important cellular constituents of the human innate and adaptive immune system and are essential for allergic reactions, inflammatory disorders and host defense against invading pathogens [2, 3]. They are normal residents and are numerous in mucosal tissues, skin, lung and gut, which are ports of entry for microorganisms, allergens and other substances. Pathogenic microorganisms are a worldwide public health problem and the control of infection requires the development of both Th1 and Th2 cellular immunity responses. MCs have historically been considered masters of allergy that can rapidly respond against allergens, pathogens, and tumors, including microbes and their products such as lipopolysaccharide (LPS) (Fig. 1). They express on their surface the c-kit receptor, stem cell factor (SCF) and FcεRI – the high affinity receptor for immunoglobulin E (IgE) (Kd = 10−10 M), which is constitutively expressed [4].
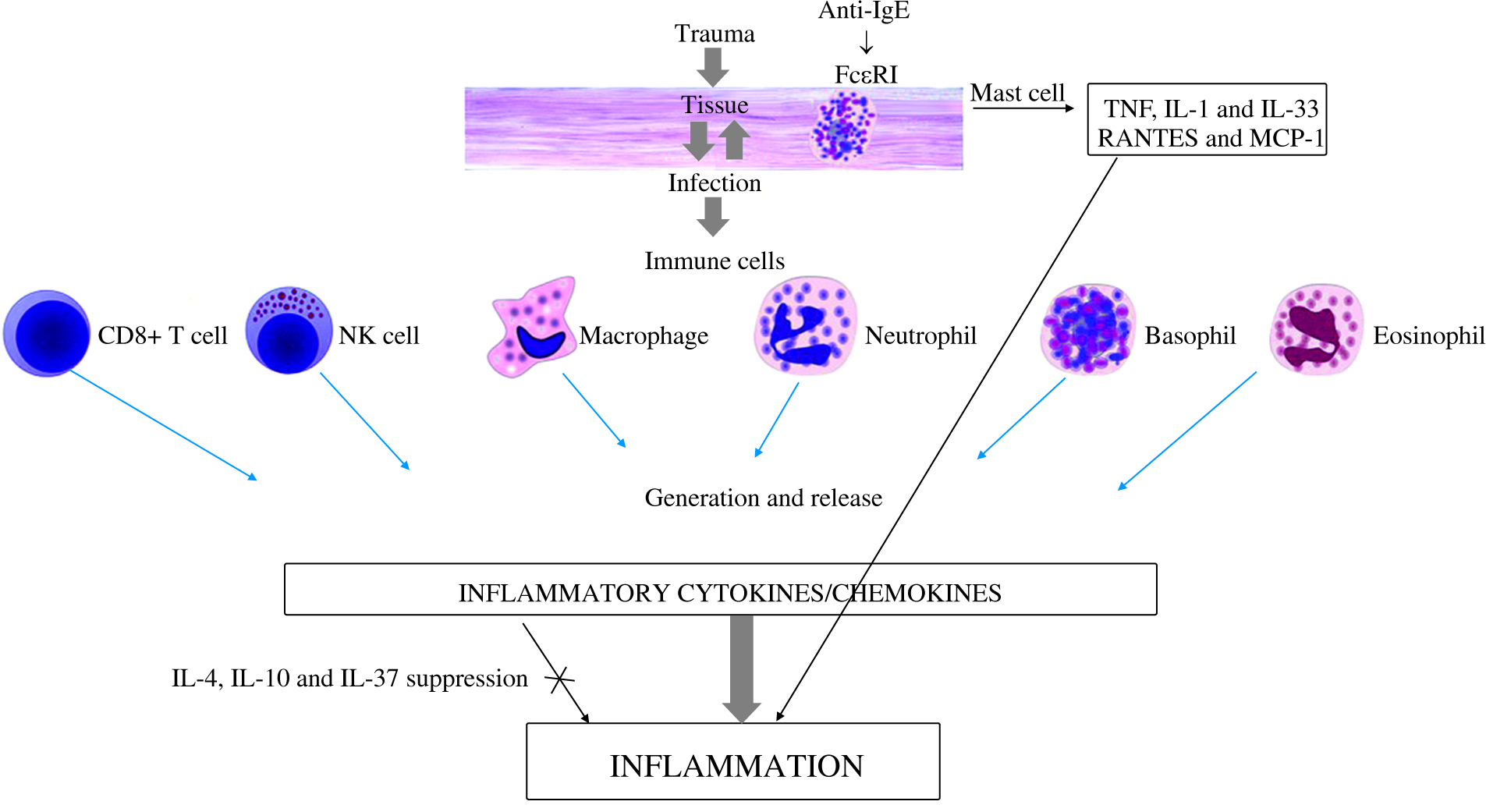
Fig. 1
Inflammatory response exerted by cytokines produced by activated immune cells in the infected tissue
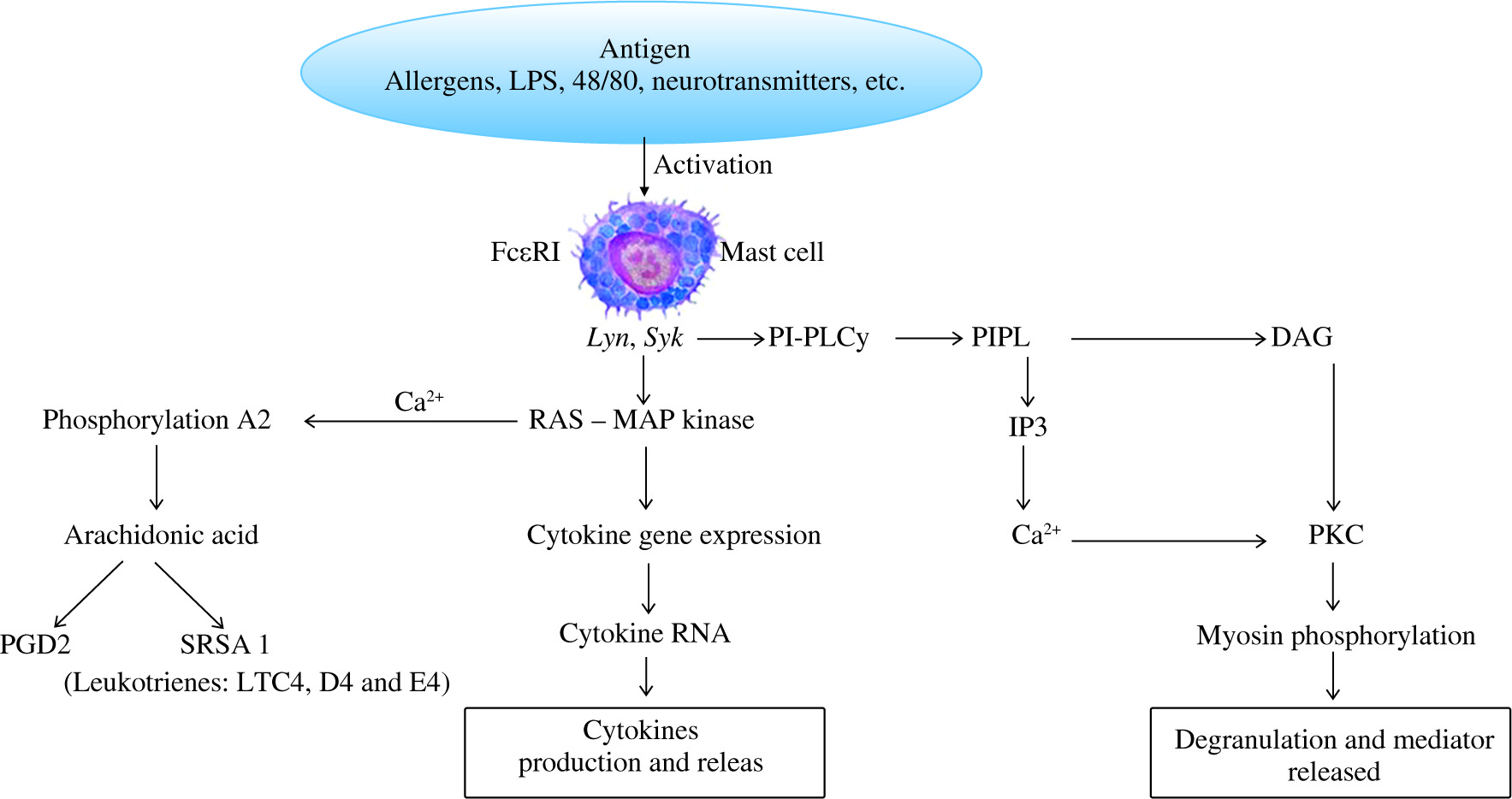
Cross-linking of bound IgE by antigen activates protein tyrosine kinases, such as Syk and Lyn, which in turn trigger activation of a mitogen-activated protein (MAP) kinase cascade and a phosphatidylinositol-specific phospholipase C (PI-PLCγ). MAP kinase acts on the MC nucleus and activates cytokine gene expression with generation of cytokine mRNA and therefore inflammatory cytokines [5, 6] (Fig. 2).
Fig. 2
Cross-linking of bound IgE by antigen(s) leads to protein tyrosine kinase activation, such as Syk and Lyn, which provoke the generation of a MAP kinase cascade resulting in release of cytokines, chemical mediators and arachidonic acid products
After infections or immunological activation in tissues, MC numbers can dramatically increase. The regulation of mast cell activation is pivotal during their immunological activity and their cytokine generation, and provides control of infections caused by microbial pathogens [7, 8].
MCs are vasodilatory, angiogenic, pro-inflammatory and neurosensitizing mediators which include in their granules histamine, heparin, kinins, neuropeptides, proteases, chymase and tryptase, which are released immediately (in seconds) [9-11]. MCs also produce newly synthesized compounds: chemokines, cytokines, growth factors, leukotrienes, prostaglandins, nitric oxide (NO), stem cell factor (SCF) and vascular endothelial growth factor (VEGF), generated and released [12]. They contribute to critical antimicrobial and regulatory functions during tissue infections [13]. MCs can participate in many of the body’s defense strategies against numerous pathogens, such as bacteria, fungi, viruses and parasites, contributing to the inflammatory response induced by these biological agents. Several lines of evidence support an important contribution for MCs in tissue microorganism clearance [14]. Various factors can modulate MC growth and survival, including several cytokines, chemokines, growth factors, some plant products, neurotransmitters and vitamins [15]. Within the tissues, mast cells are activated by microorganisms, resulting in the secretion of effector molecules. Their activation can participate in limiting microorganism replication in the local tissue, but also they can provoke vascular leakage, edema, and tissue damage [16, 17]. Some infections may provoke allergic bronchopulmonary disorders mediated by mast cells and orchestrated by CD4+ Th2-like T cells in response to persistent pulmonary microorganism allergen exposure [18]. In these diseases, there is augmentation of IgE antibodies, IgA, IgG and T-cell cytokine responses and involvement of malfunction of Treg cells.
MCs produce IL-1β and TNF, which induces vasodilation, vascular endothelial molecule expression and chemoattraction necessary for the initiation of inflammation [19, 20].
The control of infection and the battle against pathogenic microorganisms requires the activation and development of a Th1 cell immune response, as well as Th2 cells [21-23].
MCs can mediate MC recruitment after bacterial invasion, probably via multiple mechanisms, including the release of IL-1, TNF and/or IL-33 [24-27]. Activated MC-released compounds can enhance resistance to tissue infection and recruitment of other immune cells, such as lymphocytes, macrophages and neutrophils, which participate in bacterial clearance controlled by Janus kinase (JAK) 3 activation [28].
MCs and MC-derived inflammatory TNF, IL-1 and IL-33 and specific antibodies can amplify the protective adaptive immune response to infection at the site of infection [29]. The alarmin cytokine IL-33 induces protective effects in diseases by acting on neutrophils, macrophages, regulatory T cells, CD8+ cells, natural killer cells and γγ T cells, but also exacerbates the disease process by enhancing mast cell degranulation and promoting proinflammatory cytokine/chemokine generation [30]. This opposing dual effect depends on the type of infection and/or inflammation.
Since MCs also generate anti-inflammatory cytokines, such as IL-10, it is likely that this cytokine provokes an inhibitory effect on inflamed infected tissue [31, 32].
MC-derived TNF, protease and tryptase can cause microvascular damage and stimulate protease-activated receptors (PAR), leading to the inflammatory reaction [33]. Higher tryptase activity of MCs has been found in many inflammatory disorders, including infected tissue, where mast cells interact with inflamed tissue via the production of tryptase, thereby perpetuating the inflammatory response [34].
Pro-inflammatory cytokines, such as TNF-α, IL-1 family members (e.g. IL-1β, IL-18 and IL-33), TGF-β, NGF, IL-3, IL-9, and CXCL12, participate in regulating the migration, maturation, and activation of mast cells and other immune cells involved in combating infectious microorganisms [35].
c-kit mutant MC-deficient mice, such as WBB6F1-KitW/W-v, are more vulnerable to bacterial infections but, in contrast, they are more resistant to inflammatory diseases [36]. c-kit mutant mice present many immune-related disorders that can dysregulate the immune response against microbial infection in the body [37].
When immunologically activated, mast cells exhibit various degrees of anti-microbial activity. It is well established that murine mast cells possess anti-mycotic and anti-bacterial function in in vitro and in vivo treatment [38]. MCs are recruited to infection sites through chemotactic signals produced by host cells. Their characteristic of rapidly responding to invading microorganisms is an important aspect of innate immunity [39]. In fact, MCs express Fc receptors, including FcεRI, FcγRI (CD64), and FcγRIIIA (CD16A), and toll like receptors (TLRs), such as TLR-9 and TLR-3 [40], which can be activated by non-biological and biological agents, including bacteria and their derivatives. MCs can also be activated by nod-like receptors (NLR), retinoic-acid inducible gene 1-like receptors (RLR), CD48, integrins and complement receptors [41].
TLRs and the receptors for IL-1, IL-18 and IL-33 are required for defense against microbial pathogens. E. coli lipopolysaccharide activates mast cells that express toll-like receptor 2, which is critical in innate and acquired immunity [42]. TLRs in MCs mediate the production of chemical mediators and cytokines/chemokines, including IL-1β and TNF, and play a key role in innate immunity activated by infections [42]. Receptors TLR1, 2, 4, 5, 6, 10, and 11 are expressed in cell membrane; while TLR3, 7, 8, and 9 are expressed at the intracellular level. TLR microbial ligands and inflammatory cytokines/chemokines are able to activate the expression of some adhesion molecules, including VCAM-1, through stimulation of TLRs [20].
The generation of histamine by mast cells has a synergistic effect with bacterial products in the up-regulation of expression of TLR2, TLR4 and histamine H1 receptor (H1R), an effect that amplifies inflammatory responses initiated by both Gram-positive and Gram-negative bacteria infection [43].
MC activation through TLR4 leads to a strong pro-inflammatory cytokine response, but is limited to mast cell degranulation. On the other hand, activation through TLR2 induces MC degranulation and also the inflammatory cytokine response [44]. However, in some experiments on mice it has been shown that MCs are mostly activated through TLR4, rather than TLR2 after microbial infection [44].
Via the oxidative burst, mast cells and other immune cells generate hydrogen peroxide (H2O2), a reactive oxygen intermediate, one of the most important effector molecules that participate in mycocidal effects of mast cells. TNF generated and stored by MCs is capable of inducing nitric oxide synthase (NOS2) as well as expressing anti-mycotic cytokine activity [45].
MCs, upon activation by appropriate compounds such as anti-IgE and cytokines, generate TNF and nitric oxide (NO). The significance of these toxic nitrogen oxides in host defense against pathogenic microorganisms has been reported both in vitro and in vivo [44]. Therefore, NO generated by immunologically activated mast cells is one of the major antimicrobial compounds [46].
Expression of NOS2 at quite high concentration has been detected in activated MCs in mycotic infections. Moreover, the NO levels increase in tuberculosis patients [47]. Therefore, the role of mast cell NOS2 in host defense and toxic oxygen species against fungi is very important, although many aspects remain controversial.
MCs are considered a pivotal player of inflammation as they express IL-17 in inflamed tissue which is generated by the cellular source CD4(+) T cells (Th17 cells) [48, 49]. Th17 cells are involved in enhancing mucosa immunity, exerting antifungal and antibacterial function. The differentiation of Th17 cells that produce IL-17 cytokines and IL-22 is mediated by signal transducers and activators of transcription (STAT3), the deletion of which has been shown to lead to up-regulation of several Th1 cytokines [48]. Infections can trigger mast cell activation with the release of inflammatory compounds, including cytokines/chemokines, leading to the symptoms of asthma with airway edema, mucous plugging and bronchoconstriction [50]. However, the inhibition of inflammatory cytokines, such as TNF, IL-1, IL-33 and IL-17, by monoclonal antibody leads to increased vulnerability to bacterial infections [51]. STAT3, which belongs to a family of latent cytoplasmic factors, is rapidly activated by tyrosine phosphorylation in both malignant and immune cells in response to various stimuli, comprising cytokines and growth factors [52].
STAT3 is an important mediator of most immunological pathways, is ubiquitously expressed, controls numerous physiological processes, including differentiation, development, immunity, metabolism and cell proliferation, and is involved in signal transduction of several cytokines/chemokines, including IL-6, IL-10, IL-12, IL-21, IL-22, IL-23 and chemokines IP-10 (CXCL10) and RANTES (CCL5) [53]. Cytokines, including IFN gamma, induce cross-phosphorylation of JAK2 and JAK3, which results in the downstream activation of STAT3, important for cell growth [53].
A highly elevated serum IgE level, reported in STAT3-mutated hyper-IgE syndrome, implies skeletal abnormalities, pneumonia, skin and vascular diseases and mycotic infections [54].
It is well known that macrophage NOS2 is involved in host defense against M. tuberculosis even though its exact role is not yet clear. Macrophages and mast cells generate H2O2, an ROS which has mycobactericidal and mycobacteriostatic activity and inhibits pathogenic microorganism reproduction [55]. Therefore, pathogenic microorganisms are able to increase levels of intracellular reactive oxygen species (ROS). H2O2, as well as other ROS oxidants, are induced through FcεRI in macrophages and mast cells. These compounds have an important function in MC degranulation and generation of cytokines/chemokines, leukotriene secretion, and regulation of various mast cell functions. ROS may also play a relevant function in mast cell activation in allergic environmental substances [56]. H2O2, along with peroxide anions and hydroxyl radicals, promotes necrosis in certain forms of cancer. In addition, TNF generated and released by MCs also has an inhibitory effect on microbes and tumor cells [57].
As is known, TNF is one of the most potent inflammatory cytokines which mediates the activation of T cells, acting in part through the phosphorylation of NF-κB elevated in inflammatory disorders. Its suppression by pharmacological agents or anti-TNF receptor not only inhibits inflammation but also reduces the expression of other inflammatory cytokines/chemokines and inhibits Th17 cells including their products [58-60]. All of these effects influence the body’s immunity against diseases caused by pathogenic microorganisms.
Chemokines are cytokines with chemotactic activity towards inflammatory and other cells, and play a key role in the pathogenesis of inflammatory tissue [61]. Chemokine response is very important in the initial stages of bacterium-induced inflammation. The chemokine supergene family members are 8- to 10-kDa proteins inducible in a number of pathophysiological processes acting via G-protein coupled chemokine receptors that trigger different signaling pathways. Chemokines attract macrophages, lymphocytes, granulocytes and mast cells to the site of infection and regulate immune cell maturation and development. The generation and release of chemokines such as RANTES (CCL5) and MCP-1 (CCL2) from activated mast cells and macrophages are crucial steps in mast cell recruitment necessary for establishing local inflammatory responses [35].
RANTES (CCL5) is a multifunctional chemokine that has a primary role as a chemotactic factor. It is a member of the intercrine β subfamily and a C-C chemokine reported as acting as a selective chemoattractant for monocytes, T cells, eosinophils and mast cells rather than neutrophils. RANTES (CCL5), MCP-1 (CCL2) and various other chemokines play an active role in recruiting specific leukocytes into inflammatory sites and are potent chemokines involved in macrophage and mast cell activation. In addition, it has been reported that chemokines, including RANTES and MCP-1, mediate and stimulate the release of inflammatory products [35, 36].
Several chemokine receptors, especially CXCR3 protein, are preferentially expressed on mast cells in microorganism inflamed tissue [62].
Monocyte chemotactic proteins (MCP-1, MCP-2, MCP-3) and RANTES are ββchemokines capable of attracting mast cells, lymphocytes, macrophages, memory T cells and basophilic cells, but not neutrophils. CCL2/MCP-1 is one of the major and most studied members of the CC chemokines. The role of CCL2 is emerging in regulating the recruitment of inflammatory cells into infected inflamed tissue. The inhibition of MCP-1 with the corresponding specific antibody or other inhibitors may provide benefits in different clinical aspects, including inflammation caused by parasitic, bacterial and fungal diseases, and may also represent targets for diagnostic procedure, therapeutic intervention and prognostic factor.
The synthesis and expression of a plethora of chemokines, such as IL-8 (CXCL8), RANTES, MCP-1, and eotaxin, by mast cells can play an important role in inflammatory diseases triggered by microorganism infections [35, 36]. It is interesting that mast cell-deficient mice C57BL/6J-Wv/+ which have multiple defects in immune responses, are more vulnerable to bacterial and fungal infections and they have fewer inflammatory diseases, but mice lacking mast cells (W/Wv) exhibit a high percentage of mortality [35, 36]. However, the relative contribution of these chemokines to inflammatory diseases is still unclear.
In allergy, the T-helper-2 (Th2) response leads to increased generation of IL-4 and production of IgE, which binds to the receptor on the mast cell surfaces, causing the release of chemical inflammatory mediators and cytokines/chemokines. These effects lead to more hyperemic and edematous tissue which promotes microorganism adhesion, indicating that bacterial infection may exacerbate the severity of IgE-mediated allergy [63].
On the other hand, IL-4 ameliorates inflammatory disease, not only via promoting Th2 polarization, but also via direct inhibition of inflammatory cytokines/chemokines, supporting the observation that IL-4 leads to up-regulation of GATA3 mRNA and related protein in inflamed skin [64]. In several diseases, including atopic dermatitis (AD), which is mediated by mast cells with elevated specific IgE, bacterial infections are common and there is exposure of the binding sites for the bacteria in the extracellular matrix, disruptions in innate immunity, and cellular immune dysfunction, with predominant Th2 cell responses [65].
Severe inflammation, such as inflammatory bowel disease and AD, mediated by inflammatory cytokines including IL-17, is characterized by peripheral eosinophilia, and augmentation of MCs in inflamed tissue [66, 67]. In addition, viral infections also occurring in patients with AD include eczema herpeticum, coxsackium, and vaccinatum, and are occasionally complicated by secondary infection with staphylococcal or streptococcal species.
Interleukin 37
Over 35 years ago we reported that IL-1 is a family of polypeptides which induces both laboratory and clinical aspects of the acute-phase response [68].
The IL-1 cytokine family comprises several pro-inflammatory and anti-inflammatory protein molecules which orchestrate the activation of innate immunity, the first line of defense against pathogenic microorganisms. Innate immune activation, through TLR, provokes the generation of inflammatory cytokines such as IL-1, IL-18, IL-33, IL-36. These cytokines activate immune target cells and amplify the immune response, but they may also cause inflammation.
IL-37 (IL-1F7) binds the α chain of the IL-18 receptor and has five different splice variants, namely IL-1Fa, b, c, d, and e, among which IL-17b is the largest, most complete and most studied.
IL-37 down-regulates inflammation, and silencing this cytokine in monocytes results in augmentation of lipopolysaccharide and IL-1-induced cytokines [69].
IL-37, a newly discovered member of the IL-1 family, binds the IL-18Rα chain but it is not a receptor antagonist, and delivers an inhibitory signal by using TIR8. IL-37 transgenic mice are protected against LPS treatment, present less inflammation and are protected against cardiovascular, liver and infectious diseases [69]. The protection is due to the effect of IL-37 on the inhibition of inflammatory cytokines and chemokines, along with the stimulation of IL-10 which is also an anti-inflammatory cytokine. Moreover, IL-37 is also an inhibitor of adaptive immunity, since it down-regulates antigen presenting cells (APC) and therefore the activation of Treg.
IL-37 suppresses MyD88-mediated inflammatory responses and consequently inhibits NF-κB induced by TLR2 or TLR4 (Fig. 3).
Fig. 3
Activated dendritic cells release IL-1 which binds mast cell IL-1 receptor 1 and activates the transcription and translation inflammatory cytokines inhibited by IL-37
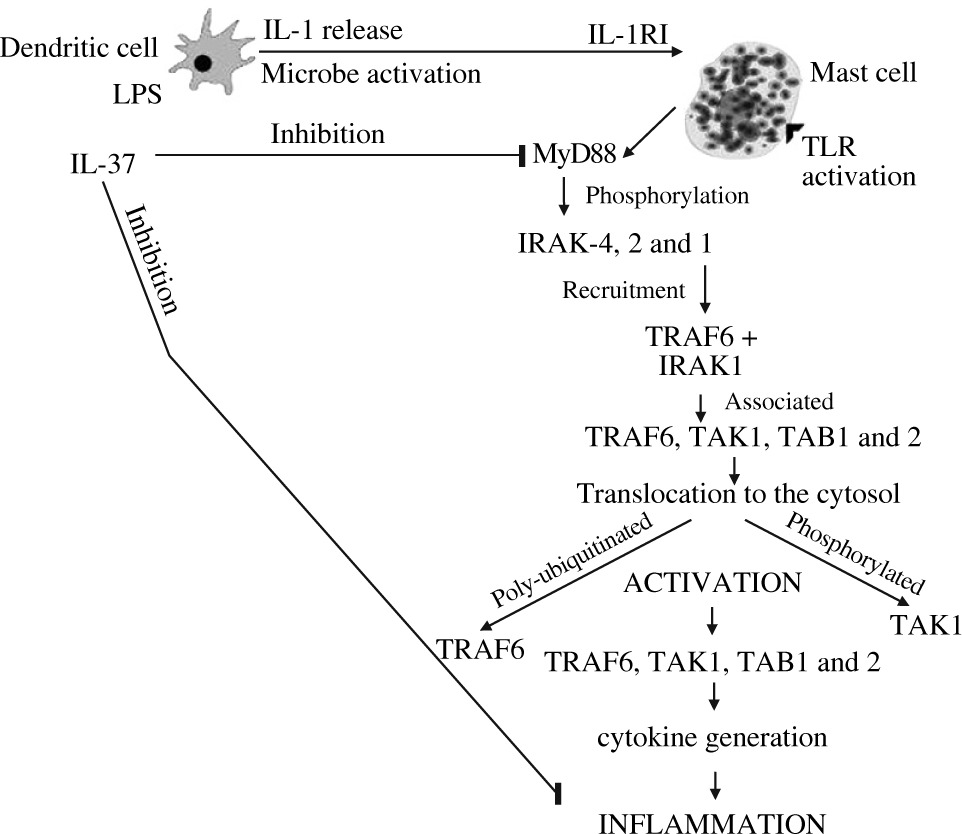
IL-37 is generated by various types of cells, such as epithelial cells, PBMC, dendritic cells, and microbe activated macrophages. This activation may release IL-1, which activates mast cells to produce inflammatory IL-6 and TNF at the site of inflammation. It has been reported that modulation of immune suppression by IL-37 inhibits both innate and adaptive immunity. Therefore, it plays a potent immunosuppressive role in the pathogenesis of inflammation by down-regulating pro-inflammatory cytokines.
IL-37 plays a pathological role in certain infections by inhibiting the production of pro-inflammatory cytokines, including IL-1 and TNF and inducing macrophages towards an M2-like phenotype [70].
Conclusions
This article summarizes our current understanding of the host immune response, with emphasis on the roles of mast cells, and the network of cytokines/chemokines in immunity against pathological microbes, offering opportunities for the design of new therapeutic interventions. However, the role of IL-37 in infection is still unclear and further research is necessary.