

Introduction
IgA is the most abundant type of immunoglobulin in the human body, and it is the second most abundant in the blood circulation, following IgG [1]. Two-thirds of all IgA is secreted in a dimeric form into the gastrointestinal and respiratory tracts, where it protects human mucosa from invading pathogens [2]. Accumulating evidence has revealed an important role of the interaction between intestinal immune system and intestinal bacterial flora in the maintenance of immune homeostasis in gut [3]. IgA-producing B cells are derived from Payer’s patches in the intestine [4]. IgA plays an important role in intestinal immunity, not only by neutralizing pathogens but also by interacting with the immune system by binding to FcαRI (CD89), which is expressed on the surfaces of B cells, T cells, and natural killer cells [5].
Selective IgA deficiency (sIgAD) is one of the most common types of immunodeficiency in humans [6]. The diagnostic criteria of sIgAD are as follows: 1) older than 4 years old, 2) serum IgA level lower than 0.07 mg/dl, 3) normal serum IgM and IgG levels, and 4) no secondary hypogammaglobinemia caused by other illnesses [7]. Patients with higher serum IgA level, but two standard deviations lower than the normal value, are classified as partial IgA deficiency (pIgAD) [6]. Previous studies have reported its prevalence as being 1 in 100-3,000 people in the United States [8] and 1 in 12,500-15,000 people in Japan [9]. Precise prevalence of pIgAD is not well investigated, but it would be much more popular than sIgAD.
Variation in the prevalence of sIgAD among countries or ethnicities suggests the existence of genetic factors. Indeed, family studies in blood donors have shown that first-degree relatives of patients with sIgAD have a 38-fold higher prevalence of sIgAD than that of controls [10]. Another study has reported a higher frequency of sIgAD coexistence between monozygotic and dizygotic twins compared with the normal population [11].
The inheritance pattern of sIgAD, however, is not clearly understood. It does not always follow Mendel’s law, as autosomal dominant, autosomal recessive, and sporadic transmission patterns have all been seen in previous family studies [12]. Some studies have reported an association between sIgAD and mutations in the transmembrane activator, and calcium-modulator and cyclophilin ligand gene [13], or a higher prevalence of a particular human leucocyte antigen haplotype [14]. However, those genetic characteristics can be only seen in a part of sIgAD patients. These facts suggest a heterogeneous genetic background among sIgAD patients.
Despite aggressive scientific investigation, the pathogenesis of sIgAD and pIgAD has not yet been fully elucidated. The most common finding in sIgAD patients is immaturity of the B cells that secrete IgA [15-17]. The fact that sIgAD can be transmitted by bone marrow transplantation indicates the existence of abnormalities in the hematopoietic stem cell series [18]. One study reported a decreased proportion of regulatory T cells in sIgAD children [19]. Other studies also have shown abnormalities in several cytokines, such as interleukin-10 and interleukin-21 in sIgAD patients [20-22].
The clinical presentation of sIgAD varies among patients, which also provides further evidence for the heterogeneity of the disease. Two-thirds of patients are asymptomatic. On the other hand, higher incidence rates of respiratory or gastrointestinal infections, allergic conditions, autoimmune diseases, some skin problems, and malignancies have been reported [23-25]. In addition, some patients develop common variable immunodeficiency (CVID) [26, 27], and suffer from anaphylactic reactions to blood derivatives [28]. A questionnaire study revealed impaired health-related quality of life in sIgAD patients compared with healthy controls because of the fear of those comorbidities [29].
Clinical presentation of pIgAD is similar to that of sIgAD. Small observation study and a case report have reported that pIgAD is related to rheumatic diseases, autoimmune diseases such as Addison’s disease, thyroiditis, and skin problems, such as dermatitis herpetiformis [30-32].
A large number of complications can be prevented by appropriate assessment and intervention. The easiest and the least invasive intervention is patient education. Patients should be informed of the possibility of severe allergic reactions to blood derivatives. We can also advise the patients to wear a medical alert bracelet, which informs healthcare providers that they should be careful when performing blood transfusions [6]. Clinicians should follow-up sIgAD or pIgAD patients regularly because the severity of IgA deficiency can fluctuate. In addition, sometimes patients can achieve complete recovery from sIgAD [33] or they can develop CVID over time [26]. Some infectious diseases can be prevented by vaccines, such as the Streptococcus pneumoniae vaccine and the Hemophilus influenzae type B vaccine. Previous studies have shown that prophylactic antibiotic therapy might be beneficial in severe cases [34].
It is difficult to predict which patients will suffer from anaphylactic reactions to blood derivatives. Higher frequencies of anti-IgA antibody detection have been reported in those who suffered anaphylactic reactions, but the specificity is low, and that measurement can result in an overestimation of high-risk patients [35]. Clinicians can prevent anaphylactic reactions by using blood products that are derived from IgA-deficient donors or that are washed with saline [36, 37]. The utility of a desensitization protocol has also been reported [38]. As shown above, early detection of sIgAD or pIgAD and adequate preventive care is important to improve clinical prognosis of such patients.
However, the heterogeneity of clinical characteristics among patients with sIgAD or pIgAD makes it exceedingly difficult to diagnose, particularly in regular primary care settings. Exclusion of other conditions is crucial for the diagnosis, which requires careful clinical consideration on the part of physician. Differential diagnoses range widely from congenital immunodeficiency, infection, and malignancies to autoimmune diseases. It is challenging for primary care physicians to consider all the possibilities. Well-known risk factors are limited to a family history of sIgAD and Caucasian race, and screening protocols for sIgAD or pIgAD have not yet been established.
Here, we present a retrospective observational study of low serum IgA patients to clarify the risk factors for pIgAD, which will provide guidance for clinicians in identifying pIgAD patients in the clinical setting. Our goal was to provide a useful pIgAD screening tool for clinicians, especially for primary care physicians, who encounter low serum IgA patients in their outpatient clinics.
Material and methods
We conducted a single-center retrospective observational study in St. Luke’s International Hospital in Tokyo, Japan. We included all adult patients who attended our outpatient clinic from April 2010 to March 2016, and whose serum IgA levels were lower than the lower limit of reference range (110 mg/dl). We excluded those who were unwilling to be recruited into a clinical study, and patients whose serum IgM or IgG levels were not within their reference ranges. For patients who had their serum IgA levels measured multiple times, only the data measured at the first detection of a low serum IgA level were used for further analyses. We compared the demographic information and laboratory measures between pIgAD patients and non-pIgAD patients. This study was approved by the ethical review board of St. Luke’s International Hospital (approval number, 16-R159).
Diagnosis of sIgAD
The primary end point of our study was the diagnosis of pIgAD. Data from all patients with low IgA levels, and normal IgM and IgG levels were reviewed retrospectively. The diagnosis of pIgAD was made by each physician based on the medical record of a patient. Those who lacked records about the cause of low serum IgA level were diagnosed by the first author (KMM). Those patients whose low levels of IgA could not be attributed to other causes of hypogammaglobinemia were diagnosed as having pIgAD. All clinical data were collected through the electronic chart system.
Data collection
We also collected the patients’ demographic information and other laboratory results obtained at the time closest to the measurement of serum IgA. The demographic information collected included age and gender. The laboratory results collected included complete blood counts, serum protein levels, renal function tests, liver function tests, serum immunoglobulin levels, and serum complement levels. All data were extracted automatically from the electronic medical records. We categorized age into two group, i.e., those who were younger than 60 years, and those who were 60 years and older. White blood cell counts were divided into two groups, such as less than 10,000/µl, and 10,000/µl or more. In terms of hemoglobin level, we divided patients into three groups, namely, those with hemoglobin levels less than 10.0 g/dl, 10.0-15.0 g/dl, or more than 15.0 g/dl.
Statistical analysis
Univariate analysis was performed by the χ2 test for categorical variables and Student’s t-test for continuous variables. Multivariate analysis was performed by logistic regression analysis to calculate odds ratios and their 95% confidence intervals. The explanatory variables were selected based on the results of the univariate analysis and clinical judgement. Those with p values less than 0.2 in the univariate analysis were considered as potential explanatory variables in the multivariate analysis. To impute missing data for total protein, albumin, creatinine, blood urea nitrogen, IgM, and IgG levels, we applied the multiple imputation method with 10 imputation sets based on age and gender. We also performed receiver operating characteristic (ROC) curve analysis to validate the model used in our multivariate analysis. The dependent variables selected were the ones used in the multivariate analysis. All the analyses were performed using Stata/MP 14 (StataCorp LLC, TX, USA).
Results
Patients’ demographics
In total, 259 patients were recruited into our study. Their mean age was 50.2 years old (standard deviation, 15.6), and the number of female patients was 221 (85.3%).
Diagnosis of sIgAD
Based on the retrospective chart review, 181 (69.9%) patients were diagnosed as having pIgAD. We classified 78 patients in non-pIgAD group because they had drug-induced hypogammaglobinemia (33 patients), hematologic neoplasms (25 patients), renal loss due to proteinuria (10 patients), solid neoplasms (9 patients), or cryoglobulinemia (1 patient) (Table 1).
Univariate analysis
The x2 test revealed that there were significantly more women in the pIgAD group than in the non-pIgAD group (p < 0.001). Student’s t-test showed statistically significant differences between the two groups in terms of white blood cell counts, hemoglobin levels, serum protein levels, and renal function tests (Table 2). In the pIgAD group, the patients were younger; their white blood cell, hemoglobin, and serum protein levels were higher, and their renal function was better than in the non-pIgAD group.
Table 2
Univariate analysis
Multivariate analysis
We conducted a logistic regression analysis to compare the pIgAD group and the non-pIgAD group (Table 3). It revealed that female gender (odds ratio (OR): 5.43; 95% confidence interval (CI): 4.88-5.98), low white blood cell count (less than 10,000/µl) (OR: 7.59; 95% CI: 6.89-8.45), and normal hemoglobin level (10-15 g/dl) (OR: 3.58; 95% CI: 3.37-3.82) were predictive factors of the pIgAD diagnosis. After estimating any missing values using the multiple imputation method, the analysis object increased from 195 to 257, which revealed that younger age (younger than 60 years old) (OR: 2.36; 95% CI: 2.25-2.48) was also a significant prognostic factor.
Table 3
Multivariate analysis
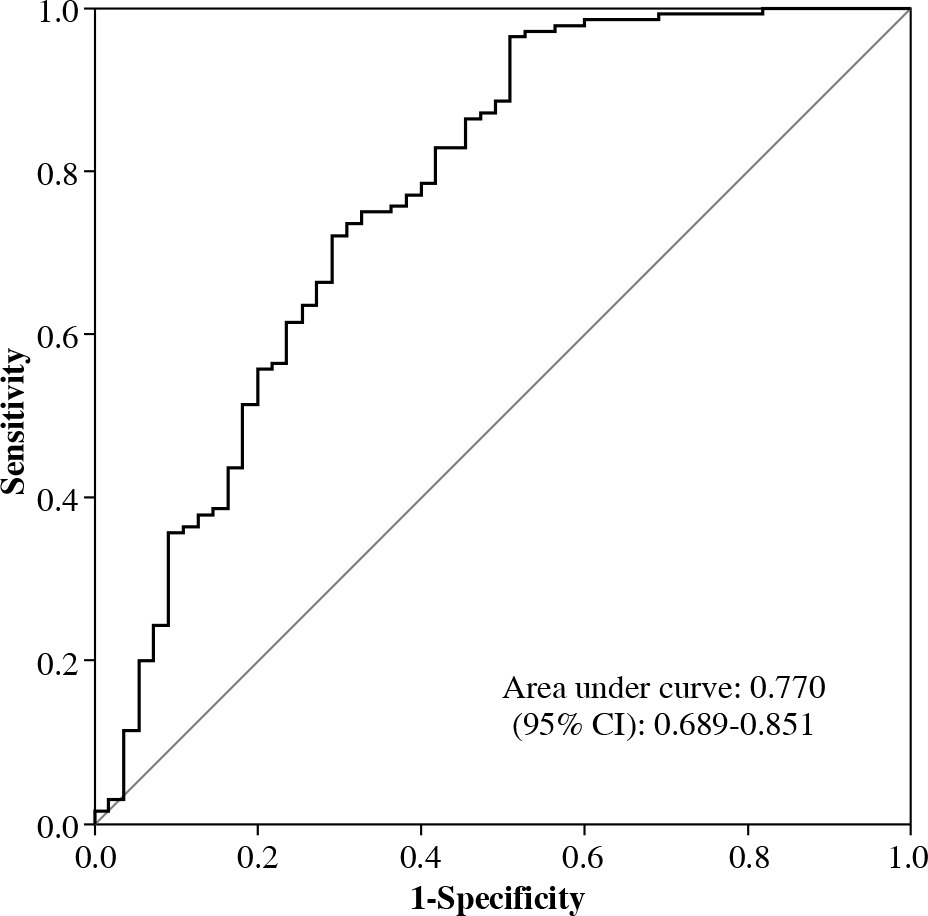
ROC curve analysis
As shown in Figure 1, the area under the curve (AUC) was 0.770 (95% CI: 0.689-0.851), which indicated that our statistical model was moderately accurate.
Discussion
Our analysis showed that risk factors for pIgAD in low serum IgA patients were as follows: female gender, age younger than 60 years old, white blood cell count lower than 10,000/µl, and hemoglobin level between 10.0-15.0 g/dl. ROC analysis validated our statistical model. When clinicians find such characteristics in low serum IgA patients, we recommend they attempt to rule out other causes of low serum IgA.
There have been some case-control studies that have compared the clinical features of sIgAD patients with those of controls [23, 25]. However, none of these studies have focused on the diagnosis of sIgAD or pIgAD. Our study is the first investigation targeting low serum IgA patients to emphasize pIgAD diagnosis. This study is characterized by a diagnostic flow like that in the real-world clinical setting. In many cases, accidental detection of low serum IgA is the first piece of evidence suggesting the presence of pIgAD, which is the reason we specifically focused on patients with low serum IgA levels. Thus, this report provides clinicians with a practical guide to screening for pIgAD, including sIgAD, when they encounter low serum IgA patients in their outpatient clinics.
Although the prevalence of pIgAD is relatively higher than that of other types of immunodeficiency, it is thought that a number of patients are overlooked due to the lack of an established screening method and the difficulty of diagnosis owing to the heterogeneity of the clinical presentation of pIgAD, including sIgAD. On the other hand, previous studies have shown that sIgAD patients suffer from infectious and allergic sequelae, and impaired health-related quality of life [29]. Our study will help sIgAD and pIgAD patients who have been underdiagnosed and will assist primary care physicians who must screen many patients in their busy daily practice. The quality of health of sIgAD and pIgAD patients can be improved by early detection followed by appropriate preventive interventions, including patient education, close monitoring, vaccination, and careful blood transfusions.
The lower incidence of pIgAD in males and older patients may be due to the higher prevalence rates of other illnesses that exclude the diagnosis of pIgAD, such as acute myelogenous leukemia, malignant lymphoma, multiple myeloma, and some types of solid neoplasms. Also, it is reasonable to use abnormal white blood cell counts and hemoglobin levels to exclude diagnosis of pIgAD because they are the signs of hematologic malignancies, which preclude a diagnosis of pIgAD.
In our study, univariate analysis showed that serum protein levels were higher and renal function was better in pIgAD patients than in non-pIgAD patients, although that finding was not supported by the multivariate analysis. Serum protein levels might be lower in non-pIgAD patients because some debilitating inflammatory diseases such as autoimmune diseases and malignancies can cause both low serum protein level and secondary hypogammaglobulinemia. Low renal function among non-pIgAD patients might be due to those who are classified in non-pIgAD group because of their renal IgA loss via proteinuria.
We also compared the incidence of comorbidities related to pIgAD, such as fever, respiratory infections, gastrointestinal symptoms, arthralgia, or skin rash, in the pIgAD group and the non-pIgAD group. However, we could not detect any significant differences between the two groups (data not shown). We believe this was because the serum IgA levels were low in both groups. Regardless of the cause of low serum IgA, the clinical presentation of the patients was influenced by their serum IgA level. We concluded that it is difficult to detect the clinical features of pIgAD patients shown in previous case-controlled studies, such as recurrent infections or higher incidence of allergic and autoimmune diseases, because the serum IgA levels were low not only among the pIgAD patients but also among the controls in our study design.
Conclusions
The limitations of this study are mostly attributed to its retrospective design. Differences in comorbidities or follow-up periods among the subjects might bias the pIgAD diagnosis; more comorbidities or longer follow-up times lead to more thorough medical evaluations, thereby increasing the opportunity to exclude possible differential diagnoses. The validity of the diagnoses made in this study should be confirmed by prospectively following up our patients. Validation studies should also be conducted in other hospitals, countries, and ethnic groups in the future. However, the aim of this study was to establish a screening method for pIgAD rather than a set of definitive diagnostic criteria for pIgAD. Thus, we believe our aim has been achieved to some extent in this retrospective analysis.
In addition, this study recruited only patients whose serum IgA levels were measured. We likely overlooked some pIgAD patients because their serum IgA levels were not evaluated. Further investigation is required to clarify what kind of patients should receive serum IgA measurement for pIgAD screening.