
Introduction
Silver Russell syndrome (SRS) represents a clinically and genetically heterogeneous congenital disorder and its prevalence is estimated as high as 1/100 000 population. Silver-Russell syndrome was described for the first time by Silver in 1953 and then by Russell in 1954 [1]. The epigenetic abnormality of chromosome 11p15.5 was confirmed in 30–60% patients with SRS. About 7–10% patients with SRS carry maternal uniparental disomy of chromosome 7 [1, 2]. Primary features are: prenatal growth retardation, short stature, and facial dysmorphism with triangular face, relative macrocephaly and body asymmetry. Clinical diagnosis of SRS remains difficult in spite of the constantly expended knowledge about non-specific features as cryptorchidism, cafe-au-lait skin spots and short arms. The diagnostic criteria of disease are still unclear [3]. Furthermore some facial characteristic become attenuated as the patients grows up. Finally some of the typical features of SRS overlap with those of other syndromic intrauterine growth restriction disorders [2–4].
Turner syndrome (TS) occurs in one in 2500 live-born females and is associated with characteristic features such as growth retardation and gonadal dysgenesis. Some abnormalities, such as webbed neck, lymphedema of the hands and feet, skeletal abnormalities, renal malformations and cardiac defects are less common [5]. TS is characterized by a great variability of clinical manifestations caused by a total or partial loss of X-chromosome or its structural aberrations. Approximately half of individuals with TS have X monosomy (45,X) and the rest of the patients have structural abnormalities or mosaicism such as 46,X,i(Xq), 45,X/46,XX and other variants [6]. Because of short stature, delayed puberty, increased risk of cardiovascular disease, osteoporosis and increased predisposition to develop autoimmune disorders in girls with TS, it is very important to make a diagnosis as quickly as possible [5, 6]. The coexistence of two genetic disorders is rare but not impossible and therefore we should be vigilant.
Case report
The patient, a 9-year-old girl, was born from the third pregnancy after 35 weeks of gestation by caesarean section due to intrauterine growth retardation (IUGR) with fetal distress. The birth weight was 1200 g (–3 SD), length was 38 cm (–3,2 SD), occipitofrontal head circumference was 30 cm (10 pc). Apgar scores at the fifth minute were 8. The pregnancy was complicated by viral infection in mother in the first trimester and vaginal bleeding in the third trimester. The parents were healthy, young and unrelated (mother 29 and father 32 years old). The maternal medical history was negative for smoking, drug and alcohol abuse. The family history did not show any congenital anomaly syndrome or other genetic disease, but the first pregnancy of the mother ended in miscarriage. On ultrasound examination fetal biometric parameters, nuchal translucency screening (NT) and amniotic fluid index (AFI) were normal, but the result of the first Combined Screening Test result (test PAPP-A) was abnormal, a prenatal testing was performed. The analysis of amniotic cells indicated karyotype 45,X.
At 8.5 month of age, the girl was referred to our department, with the diagnosis of TS. On physical examination: height was 64 cm (–2,5 SD ), weight was 4,1 kg (–3.9 SD), head circumference 44 cm (25–50 c). The general appearance was not compatible with TS, and there were several SRS-like somatic features such as: triangular face, relative macrocephaly, small mandible and the second and third toe syndactyly. There was also failure to thrive and feeding problems. Gastroenterological examination did not confirm any pathology of the digestive tract, and celiac disease was excluded.
Chromosome analysis on 22 metaphase cells from cultured peripheral blood lymphocytes indicated a mosaic Turner syndrome: mos45,X[14]/46,X,del(X)(p21.2)[6]. ish del(X)(p22.3p22.3)(pter-). FISH analysis excluded Y-chromosome-specific sequences. Due to the suspicion of SRS methylation-specific multiplex ligation-mediated PCR analysis (MS-MLPA) was used. Molecular study for hypomethylation of the H19 region of 11p15.5 chromosome was positive. The karyotypes of the parents were normal.
At the age of 2.5 growth and general development were retarded with the length of 81.2 cm (–4 SD), weight of 7.85 kg (–4.12 SD). Physical examination showed the characteristic abnormalities for SRS such as triangular-shaped face, frontal bossing, narrow lips, down-turned corners of the mouth, high palate, and limb asymmetry (right lower limb was shorter; Fig. 1). Gross motor delay was still visible in spite of the intensive rehabilitation with Vojta and Bobath methods. The patient started sitting, walking and talking at 13, 21, 24 months of age, respectively. She had adenoid hypertrophy and hypoacusis. The horseshoe kidney was diagnosed with the ultrasound examination of the abdominal cavity and scintigraphy of urinary tract. The heart echo scan revealed bicuspid aortic valve with no other intracardiac or extracardiac congenital malformations. MRI of the brain was normal. The biochemical and hormonal parameters did not exceed the limits of the norm except for high FSH levels (23.3 IU/l). Bone age was 2.5 at the age of 3.5 years.
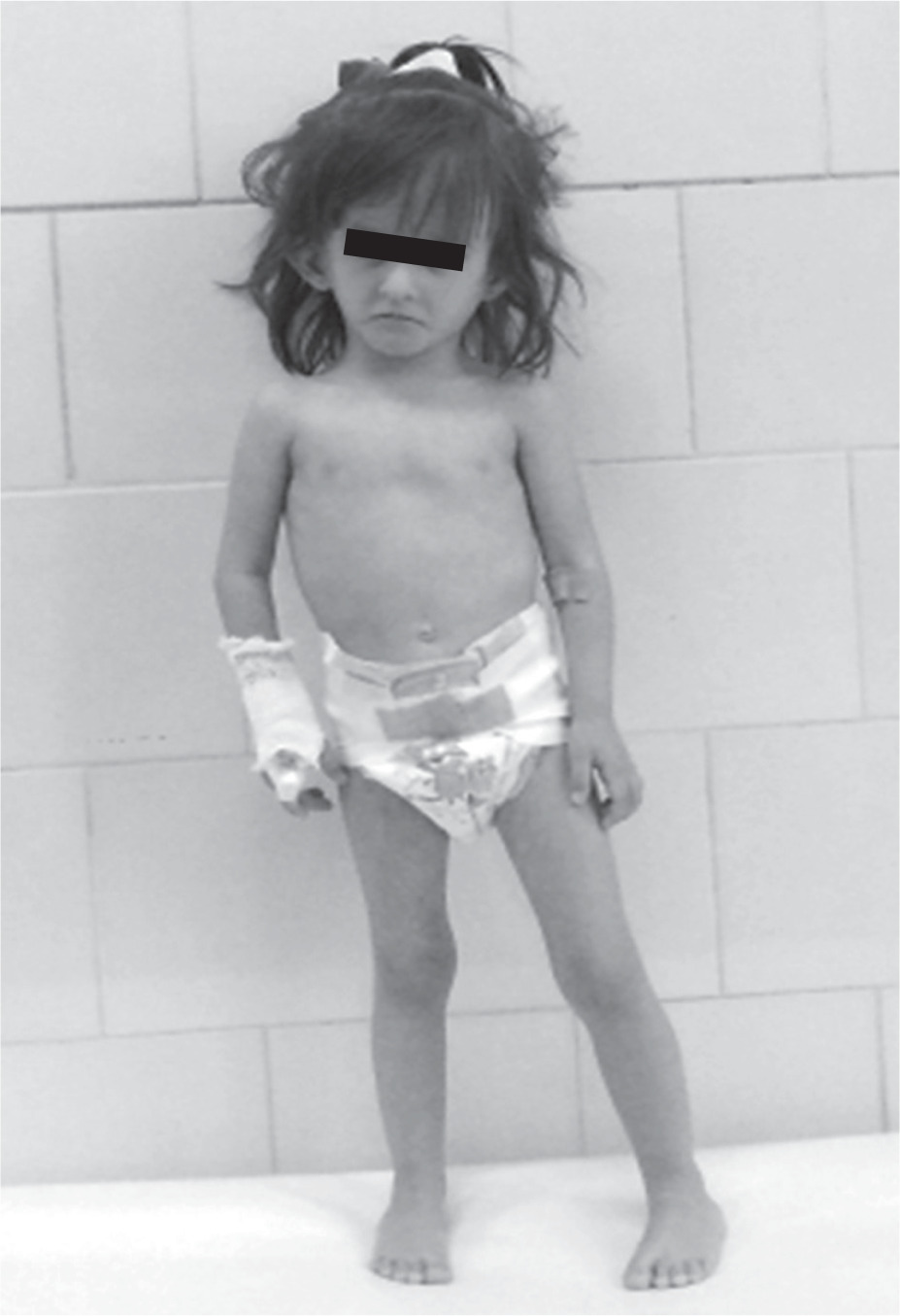
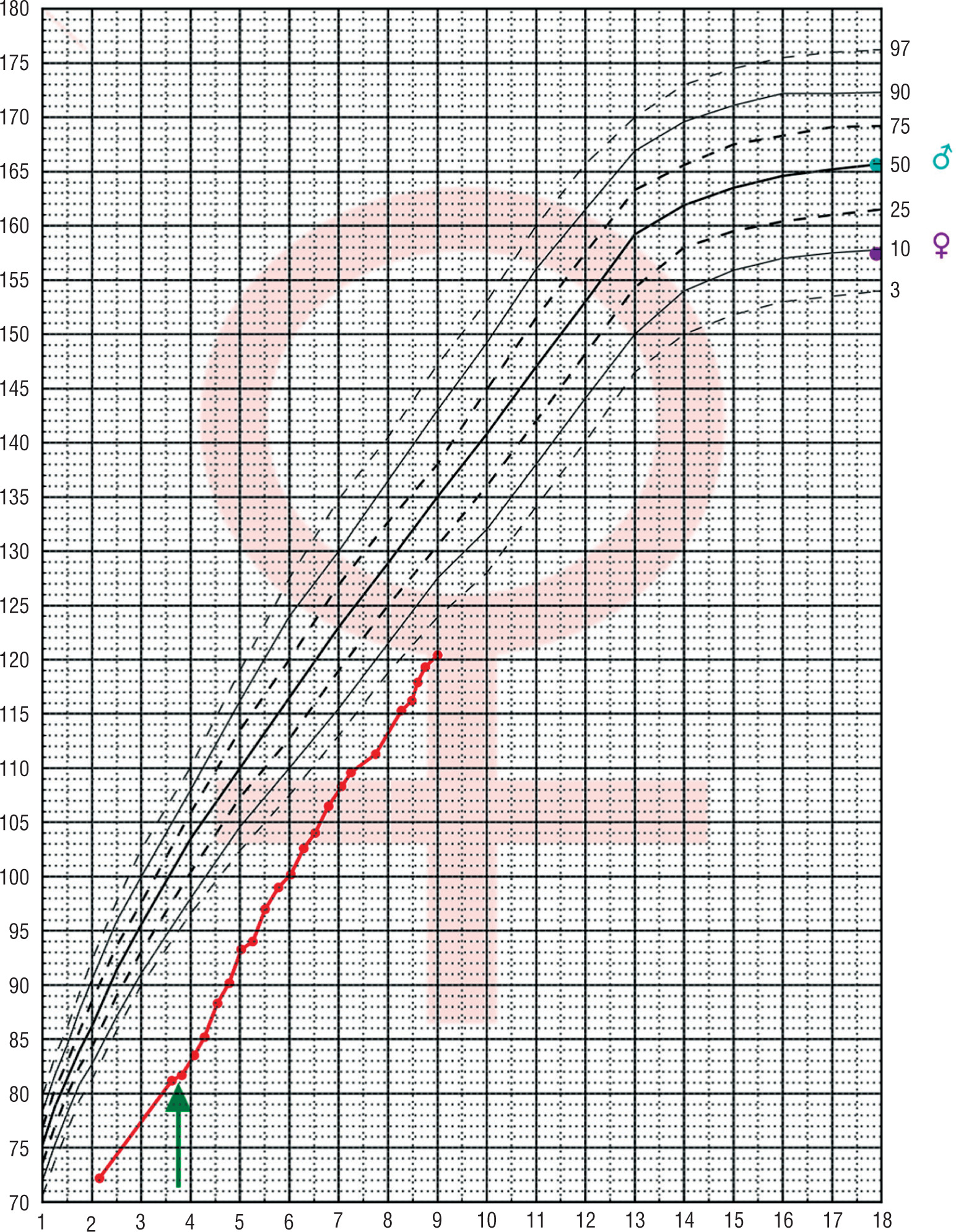
From the age of 4 the patient is treated with daily injections of growth hormone at a dose of 0.05 mg/kg of body weight without complaint. As the result the growth rate was accelerated. Before therapy growth velocity was 6 cm/year and increased to 9.3 cm/year during the first year of treatment. She has now completed the fifth year of treatment maintaining a good growth rate (8.5 cm/year; Fig. 2).
Figure 2
The growth of the patient with Silver-Russel syndrome and Turner syndrome plotted on growth chart of healthy girls.The arrow indicates the start of growth hormone treatment
The patient is making steady progress in development. Mental acuity is at lower level than a child of the same age, visual analysis and synthesis are poor. She attends the integration class and implements a lower level of education. The child is cheerful and willing to perform a test. Laboratory results are consistently correct.
Discussion
The genetic background of TS is highly variable. The most frequent occurring karyotype is 45,X. The mosaic TS occurs in only 30% of all patients with TS. There is not a clear genotype/phenotype correlation in TS, but girls with mosaic TS who have a normal cell line tend to exhibit milder phenotypes [7]. Typical phenotypic features of TS are not the same as the characteristics for the SRS but sometimes phenotypic features of both genetic disorders may overlap. Our patient’s phenotype fits the SRS spectrum better that of TS. The features association with the SRS have been described in many genetic abnormalities such as autosomal dominant or autosomal recessive and X-linked inheritance, chromosome rearrangements like maternal disomy (UPD 7) but the most cases of SRS being sporadic [2–4].
We report on the first case of SRS diagnosed as a hypomethylation of the H19 region of 11p15.5 chromosome in a mosaic TS. Blick and Gicquel have found the same hypomethylation of the H19 in SRS like in our patient [8]. The 11p15 region is also associated with the Beckwith Wiedeman syndrome. In many cases the overgrowth is asymmetric leading to hemihypertrophy. Authors hypothesized that the growth abnormality and asymmetry in both disorders might be caused by dysregulation of the same gene. H19 hypomethylation can provided from isolated asymmetry to the full clinical spectrum of SRS. In the study every patient with hypomethylation of the H19 locus had prenatal and postnatal growth retardation and asymmetry [8]. Blich et al. suggested that patients with H19 hypomethylation could have a higher risk for heart defects like atrial/ventricular septal defects. We did not observe heart disease in our patient. Coexisting two genetic disorders is rare but not impossible and unfortunately genetic tests are often negative. Li C. Chumei et al. reported a female infant who was diagnosed with mosaic TS on chromosomal basis and who manifested SRS, though molecular study for uniparental disomy 7 (UPD 7) was negative [9].
Silver-Russell syndrome is one of recognized but rare forms of early severe asymmetric intrauterine growth restrictions (IUGR) [10]. Serial ultrasound scans during the third trimester of pregnancy may be helpful in identifying short and/or asymmetric limbs. For pregnancies in which early asymmetric IUGR is identified by ultrasound, prenatal testing for UPD 7 is possible. In our case, there were no abnormalities in ultrasonography examinations and poor intrauterine growth was not observed until 18 weeks of gestation.
Silver-Russell syndrome leads at least to short stature as well as TS. We know that impaired growth in TS is caused by haploinsufficiency of SHOX (short stature homeobox gene on X chromosome). The pathogenesis of growth retardation in SRS is not fully understood and most of them have normal serum concentration of GH. The short stature in children with TS requires treatment with recombinant growth hormone in supraphysiological doses. Children with SRS do not appear to have a significantly increased incidence of neoplasia. However our patient with two genetic syndromes has to be under special and systematic control. Patients suffering from skeletal abnormalities, speech problems, learning disabilities need multispecialty intervention and individual care. All these facts require caution in assessing the final results of our medical intervention in a constantly developing patient.









