Introduction
Currently accounting for 5.8 million deaths per year, traumatic injuries are considered one of the most pressing public health problems by the World Health Organization (WHO) and are predicted to rise in significance until 2030 [1]. After polytrauma – an injury of multiple sites, life-threatening for the patient – causes of early and late mortality can be distinguished [2]. The treatment of late mortality caused by inflammatory processes is increasingly investigated as it still yields significant potential for improvement of clinical outcomes.
Upon trauma, tissue injury occurs and the immune system is activated locally and systemically by pro-inflammatory mediators [3]. Posttraumatic systemic inflammatory response syndrome (SIRS), which is associated with late mortality, can be diagnosed in up to 90% of trauma patients admitted to the intensive care unit [4]. More specifically and according to the widely accepted two-hit-model, SIRS can be caused by injuries directly related to trauma (“first hits”) or by secondary stress such as infection, ischemia/reperfusion and surgery (“second hits”) and can ultimately lead to multi-organ-failure (MOF) [5]. In a protective attempt to restore immunologic balance, hyperinflammation is countered by an anti-inflammatory mechanism called compensatory anti-inflammatory response syndrome (CARS) [6].
At the cellular level, posttraumatic pro-inflammation is predominately driven by the innate immune system via tumor necrosis factor α (TNF-α), interleukin (IL)-1β, IL-6, IL-8, neutrophil activating peptide (NAF), macrophage migratory factor (MMF), high mobility group protein-1 (HMG-1), IL-12, IL-18, etc. [7]. During the posttraumatic compensatory anti-inflammatory response, counter-inflammatory cytokines such as IL-4, IL-10, IL-13 are released [7] and adaptive immunity mechanisms are altered [7].
CD4+ regulatory T cells (CD4+ Tregs), a special T-helper-cell subset, are considered to play a key role in the trauma-related immune disturbance [8]. It has been shown that they are activated posttraumatically [9, 10] and their function is enhanced following injury [9], whereas the mechanisms leading to posttraumatic CD4+ Treg activation are not fully understood. Classically, CD4+ Tregs were understood to exhibit a protective function by suppressing pro-inflammatory T cell activity via anti-inflammatory cytokines [IL-10, IL-35, transforming growth factor β (TGF-β)] and direct cell-cell contact [11]. This exclusively protective anti-inflammatory role is currently being reevaluated, as CD4+ Tregs were observed to produce IL-17 and plasticity of CD4+ Tregs towards proinflammatory acting Th17 cells could be observed [12].
The role of platelets as a potential interaction partner for CD4+ Tregs was analyzed due to their complex, clinically relevant immunologic features, especially the capability to modulate adaptive immunity by influencing T-helper cell function [13].
As observed in mechanistic in vitro stimulation studies and considered relevant in inflammatory and neoplastic settings, platelets and CD4+ Tregs can interact [14, 15]. Our group followed the hypothesis of a possible interaction of platelets and CD4+ Tregs in the posttraumatic setting and demonstrated in a murine depletion model that posttraumatic activation of CD4+ Tregs indeed is platelet-dependent following burn injury [16]. Furthermore, we recently observed that paracrine interactions via TNF-R2- and TLR4-dependent pathways have an effect on the interaction of platelets and CD4+ Tregs following trauma [17].
In other immunologic settings, e.g. hepatic ischemia-reperfusion injury, it has been shown that CD4+ T cells and platelets communicate in a direct cell-cell contact manner via CD40L and P-selectin-dependent pathways [18, 19]. It remained unclear, whether these molecules modulate the reciprocal activation of CD4+ Tregs and platelets following trauma-induced injury.
CD40L (CD154) and its receptor, CD40, are costimulatory molecules of the TNF (receptor) superfamily [20]. CD40L is predominately expressed on activated T cells as well as the CD4+ Treg subset [21-23], while CD40 is expressed constitutively on a variety of cells including platelets [19, 24]. CD40 and CD40L are crucial for the development and homeostasis of CD4+ Tregs [22, 25]. In a CD40-CD40L-dependent interaction of CD4+ T cells and platelets, activated T cells can trigger platelet activation via membrane-bound CD40L and, vice versa, platelets can recruit T cells [19].
The adhesion molecule P-selectin (CD62P, P-sel) belongs to the Ca2+-dependent lectin family and ligates to P-selectin glycoprotein ligand-1 (PSGL-1). With the ligand-receptor pair being pathomechanistically relevant in different inflammatory medical conditions [26], PSGL-1 is expressed on T cells [27], while P-selectin is translocated from the platelets’ α-granules to the external cellular membrane upon platelet activation and thus is considered a phenotypic and functional activation marker on platelets [28]. Furthermore, P-selectin expression has been shown to be significantly higher than on CD4+ non-Tregs, and in vitro and in vivo studies indicated P-selectin to be a critical molecule for CD4+ Treg differentiation and function [29].
Trauma-induced injury leads to reciprocal activation of CD4+ Tregs and platelets, as shown in previous studies by our group [16]. The mechanisms of this cell interaction remained unclear. In this screening study, we utilized CD40L- and P-selectin knockout mice in a burn injury model to investigate the potential influence of molecules known to be involved in cell-cell contact signaling of CD4+ Tregs and platelets following trauma. Indeed, our findings indicate that the cell-cell contact molecules CD40L and P-selectin play a substantial role in the posttraumatic activation of both CD4+ Tregs and platelets. Upon disruption of P-selectin- or CD40L-dependent pathways, the posttraumatic activation of CD4+ Tregs is diminished and the platelet-dependent hemostatic function is impaired. Our findings provide further understanding of the potential mechanisms of the interaction of CD4+ Tregs and platelets following trauma.
Material and methods
Animals
Disruption of the relevant pathways was mimicked using knockout mice, male B6.12957-SelptmBay/J (P-Sel –/–) and male B6.12952-Cd40lgtm1lmx/g (CD40L –/–) from Charles River Laboratories (Sulzfeld Germany); male C57BL/6N mice from Charles River Laboratories (Sulzfeld Germany) were used as controls. The animals (22-26 g) were acclimatized (1 week) and kept in the Center for Preclinical Studies (ZPF) of Klinikum rechts der Isar, Munich, Germany. A 12 h light/dark regimen and fully standardized supply (feeding and water ad libitum, controlled temperature and humidity) were provided in the virus antibody-free facility. The experimental protocols fully met the standards of the EU regulations on protection of animals. The study was approved by the responsible committee on the Ethics of Animal Experiments of the Technical University Munich (Regierung von Oberbayern, Munich, Germany, Permit Number: 164-12).
Reagents
Preparation and processing of the cells was performed adhering to the processing and staining protocol established in the proof-of-principle experiments of our group by Bergmann et al. [16] to ensure comparability of the data. Cells from draining lymph nodes were suspended in a complete-5 (C5) culture medium prepared from RPMI 1640, penicillin/streptomycin, L-glutamine 200 mM, HEPES buffer solution (1 M), 2-mercaptoethanol and 5% fetal bovine serum (FBS; all purchased from Fisher Scientific GmbH, GIBCO, Schwerte, Germany) and minimal essential medium (MEM; Sigma-Aldrich Chemical Company, St. Louis, MO). Cell staining was performed in phosphate buffer in the presence of ArgHCl (PBA) buffer solution composed of phosphate-buffered saline (PBS), albumin from bovine serum (BSA) and sodium azide (1000 : 10 : 1), all from Sigma-Aldrich (St. Louis, MO). Fixation and permeabilization were conducted using 0.15% paraformaldehyde (PFA) and methanol, from Carl ROTH (Karlsruhe, Germany).
Non-specific binding was prevented employing Fc-block reagent (purified anti-mouse CD16/32 (93)) from BioLegend (San Diego, CA). Staining of the cell surface was performed with anti-CD4 mAb (GK1.5) from Miltenyi Biotec (Bergisch Gladbach, Germany). Identification of FoxP3+ cells was achieved employing anti-FoxP3 mAb (FJK-16s) from eBioscience (San Diego, CA). Intracellular signaling molecules were stained with primary antibodies including specific anti-PKC-δ (P632), anti-p-PKC-δ (Thr 538), anti-ZAP-70 (99F2) and anti-p-ZAP-70 (Tyr 493) purchased from Cell Signaling Technology (Danvers, MA). To enable detection, the Alexa Fluor 555 conjugated F(ab)'2 fragment of goat anti-rabbit IgG from Life Technologies (Carlsbad, CA) was employed as a secondary antibody.
Mouse injury model
Anesthesia of the mice was performed with intraperitoneal application of 200 mg/kg dosed ketamine (Ursotamin 100 mg/ml, Serumwerk Bernburg AG, Bernburg, Germany) and 10 mg/kg dosed xylazine (Xylazin 2%, Ceva Tiergesundheit GmbH, Duüsseldorf, Germany). The dorsum fur was shaved and the animals were placed in a plastic apparatus exposing 25% of the total body surface. By immersing the exposed part of the dorsum in hot water (90°C) for 9 s, a third-degree burn injury was induced. Sham-treated mice followed the same protocol without contacting the hot water. After the treatment, the animals were rehydrated by intraperitoneal injection of 0.9% saline (1 ml). All mice remained narcotized under observation and were euthanized by CO2 asphyxiation after 1 h. Previous studies identified this time point as kinetically optimal for phospho-flow cytometric investigations, as the peak of fluorescence intensity of (p-)ZAP-70 and (p-)PKC-δ is reached at 60 min after trauma [10]. Animals having deceased prior to this time point were excluded from flow cytometric measurements.
Blood and lymph node cell preparation
Blood extraction for thromboelastometric analysis was performed via cardiac puncturing using a 14 gauge needle mounted with a syringe prepared with 3.1% trisodium citrate (blood/citrate ratio 10 : 1). Draining inguinal, axillary and brachial lymph nodes were harvested. Cell suspensions were centrifugated and resuspended in C5 before being strained with stainless steel meshes and sterile filters with 70 µm micron pores. The cell isolations were transduced to 96-well plates for the staining protocol and fixed with 0.15% PFA.
Staining protocol and flow cytometry
After washing the cells with PBA, permeabilization was performed with ice-cold methanol for 10 min. Fc-block reagent was added to the suspension (15 min). To identify CD4+FoxP3+ Treg cells, staining antibodies – APC-labeled anti-CD4 and FITC-labeled anti-FoxP3 (both rat anti-mouse) – were employed. For the staining of intracellular TCR signaling molecules as a tool to measure early (1 h) CD4+ Treg activation, the cell suspension was incubated with either anti-ZAP-70, anti-pZAP-70, anti-PKC-δθ or anti-pPKC-δ (all rabbit anti-mouse primary antibodies, unconjugated). After a PBA washing cycle, the cells were incubated with the Alexa Fluor 555-labeled (F(ab)'2 fragment of goat anti-rabbit IgG (secondary antibody) for 30 min at room temperature in darkness). Furthermore, negative controls to assess the impact of autofluorescence were generated, following the same protocol without undergoing incubation with staining antibodies. After another washing cycle, fixation was ensured with 0.3% PFA for a minimum of 1 h. For analysis, cells were washed and resuspended in PBS. Phospho-flow cytometry (measurement of median fluorescence intensity of the stained signaling molecules) was performed using MACSQuant from Miltenyi Biotec GmbH (Bergisch Gladbach, Germany) with a flow rate of 10,000 events/second. Beads from Miltenyi Biotec GmbH (Bergisch Gladbach, Germany) were employed to calibrate the device each time. Analysis of the phospho-flow cytometry output data was conducted with FlowJo v.10 (FlowJo, LLC, Ashland, Oregon). Identification of CD4+ Tregs was ensured by gating for live cells, singlets, CD4+ positivity and foxp3 positivity. Microsoft Excel v16.23 (Microsoft Corporation, Redmond, Washington) was used for data processing. A relative fluorescence intensity (RFI) adjustment was performed by calibrating the median fluorescence (MFI) of the samples with the MFI of each animal’s unstained control, to exclude influences from autofluorescence based on the RFI method presented by Hanschen et al. [10, 30].
Rotational thromboelastometry
To analyze the hemostatic function of platelets, rotational thromboelastometry analysis was performed using ROTEM delta (TEM International GmbH, Munich, Germany). In ROTEM Cup&Pin mini sets from TEM International GmbH (Munich, Germany), 7 µl of star-tem for re-calcification and 7 µl of ex-tem substance were added to 105 µl of full mouse blood combined with 3.1% trisodium citrate in a ratio of 10 : 1. The analyzed ROTEM parameters include clotting time (CT), clot formation time (CFT), alpha angle and maximum clot firmness (MCF). The CT describes the time until a clot formation of 2 mm amplitude is detected. The CFT defines the latency from the initiation of clotting until reaching a clot firmness of 20 mm in amplitude, while the alpha angle defines the angle of a tangent from the initiation of the clotting to the slope of the coagulation graph. Together, alpha angle and CFT are indicators of clot formation velocity. The MCF is defined as the maximum width of the coagulation graph indicating maximum firmness of the clot, serving as an indicator of the platelet’s clotting quality and strength [31].
Experimental protocols
Effect of CD40L and P-selectin on activation of CD4+ Tregs after trauma
Experiments were conducted according to the protocols described above. One hour following third degree burn injury, the experimental animals’ draining lymph node cells were harvested. Employing phospho-flow cytometry, expression and phosphorylation of intracellular TCR signaling molecules were measured to determine posttraumatic activation of CD4+ Tregs. A male C57BL/6N WT control group was compared to a CD40L- and P-selectin-deficient group respectively.
Effect of CD40L and P-selectin on functional parameters of platelets after trauma
Rotational thromboelastometry (ROTEM delta) was performed on blood samples one hour following third degree burn injury using the EXTEM protocol to examine the effects of trauma on platelet activation and function. A male C57BL/6N WT control group was compared to a CD40L- and P-selectin-deficient group respectively, to clarify the mechanisms of the previously observed reciprocal posttraumatic activation of CD4+ Tregs and platelets.
Statistical analysis
Relevant comparisons between the experimental subgroups were identified and for the relevant comparisons, the Mann-Whitney U-test was applied using GraphPad Prism Software version 6.05 from GraphPad Software, Inc. (La Jolla, CA). Figures were created using GraphPad Prism Software version 6.05 from GraphPad Software, Inc. (La Jolla, CA) and MS Office v16.23 (Microsoft Corporation, Redmond, Washington). Box plots show the median ± Q1 and Q3 values, whiskers represent the minimum and maximum value of each data set. P < 0.05 was considered significant.
Results
Phenotypes of CD40L and P-selectin knockout models allow for assessment of CD4+ Treg function in the murine trauma model
We phenotyped the cell populations in the lymph node tissues to ensure viability of the knockout models. In agreement with the literature [32], in the CD40L-KO the overall lymph node cellularity was significantly reduced to 29% (p < 0.001), the CD4+ Treg count was significantly reduced to 30% (p < 0.001) compared to WT controls, but the percentage of the CD4+ Treg subpopulation remained similar, with a difference of only 0.5% (data not shown). In the P-selectin deficiency – in agreement with the literature [29] – we measured a significantly, 1.59-fold higher total cell count (p < 0.001), a significantly, 1.68-fold higher CD4+ Treg count (p < 0.01) and a percentage of the CD4+ Treg subpopulation in the range of wild-type animals with a difference of only 2.4% (data not shown). These findings show that CD4+ Tregs – the cell type of interest in this study – are still produced in sufficient quantities for measurement in the knockout model.
Trauma induces early activation of CD4+ Tregs
We assessed the activation of CD4+ Tregs by performing phospho-flow cytometry measuring the early posttraumatic expression and phosphorylation of signaling molecules representative for CD4+ Treg activation – (p)ZAP-70, (p)PKC-δ – as previously established [10, 16, 30]. ZAP-70 and its activated phosphorylated version (pZAP-70) are important downstream signaling molecules following TCR activation leading to T cell activation [33], while PKC-δ-phosphorylation can be triggered by both TCR-dependent and TCR-independent mechanisms [30]. In Figure 1A, we show the gating strategy used for identifying FoxP3+ CD4+ T cells. In lymph node based CD4+ Tregs of WT animals, we observed biologically significant activation upon trauma in this study when comparing the sham-treated group with the burn-intervention group with (△= delta median RFI of compared groups) △ZAP-70: 0.06 (p = 0.13), △pZAP-70: 0.12 (p < 0.05), △PKC-δ: 0.10 (p < 0.05), △pPKC-δ: 0.06 (p < 0.05) (Figs. 2 and 3). These effects were not observed in the spleen in previous studies [10, 16, 17]. In Figure 1B we show a histogram representing the difference in fluorescence signal strength between sham and burn-treated animals.
Fig. 1
Phospho-flow cytometry allows for the analysis of posttraumatic CD4+ Treg activation. Phospho-flow cytometric measurement of expression and phosphorylation of intracellular signaling molecules, ZAP-70 and PKC-θ, in CD4+ FoxP3+ T cells (CD4+ Tregs). A) Gating strategy to identify CD4+ Tregs in previously identified CD4+T cells. B) Representative histogram displaying posttraumatic activation of CD4+ Tregs measured by expression of PKC-θ in CD4+ Tregs in sham-treated (light-grey line and filling) and burn-treated animals (dark-grey line and filling)
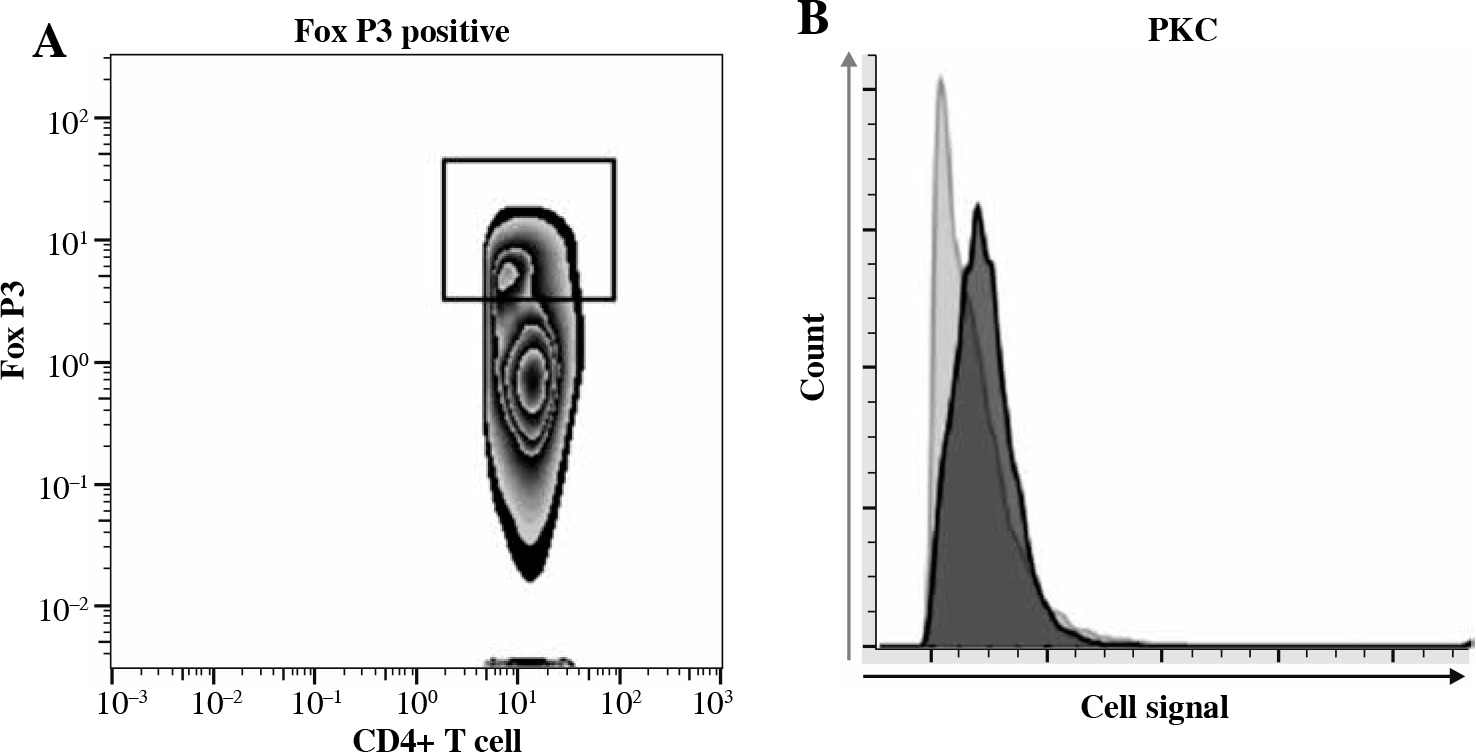
Fig. 2
Baseline activity, posttraumatic activation and posttraumatic activity in lymph node based CD4+ Tregs are lower in CD40L-KO animals than in WT animals. The baseline activity measurement of lymph node based CD4+ Tregs is significantly lower in CD40L-KO animals than in WT animals. Trauma leads to a significant posttraumatic increase of the phospho- flow cytometric signal of CD4+ Tregs in WT animals (significant for pZAP, PKC, pPKC). In contrast, the posttraumatic phospho-flow cytometric signal is diminished in CD40L-KO animals and settles at a significantly lower level compared to WT animals. Boxes: Median ± Q1 and Q3, whiskers: min. and max value of each data set, n = 6-15 per group, *p < 0.05, **p < 0.01, ***p < 0.001
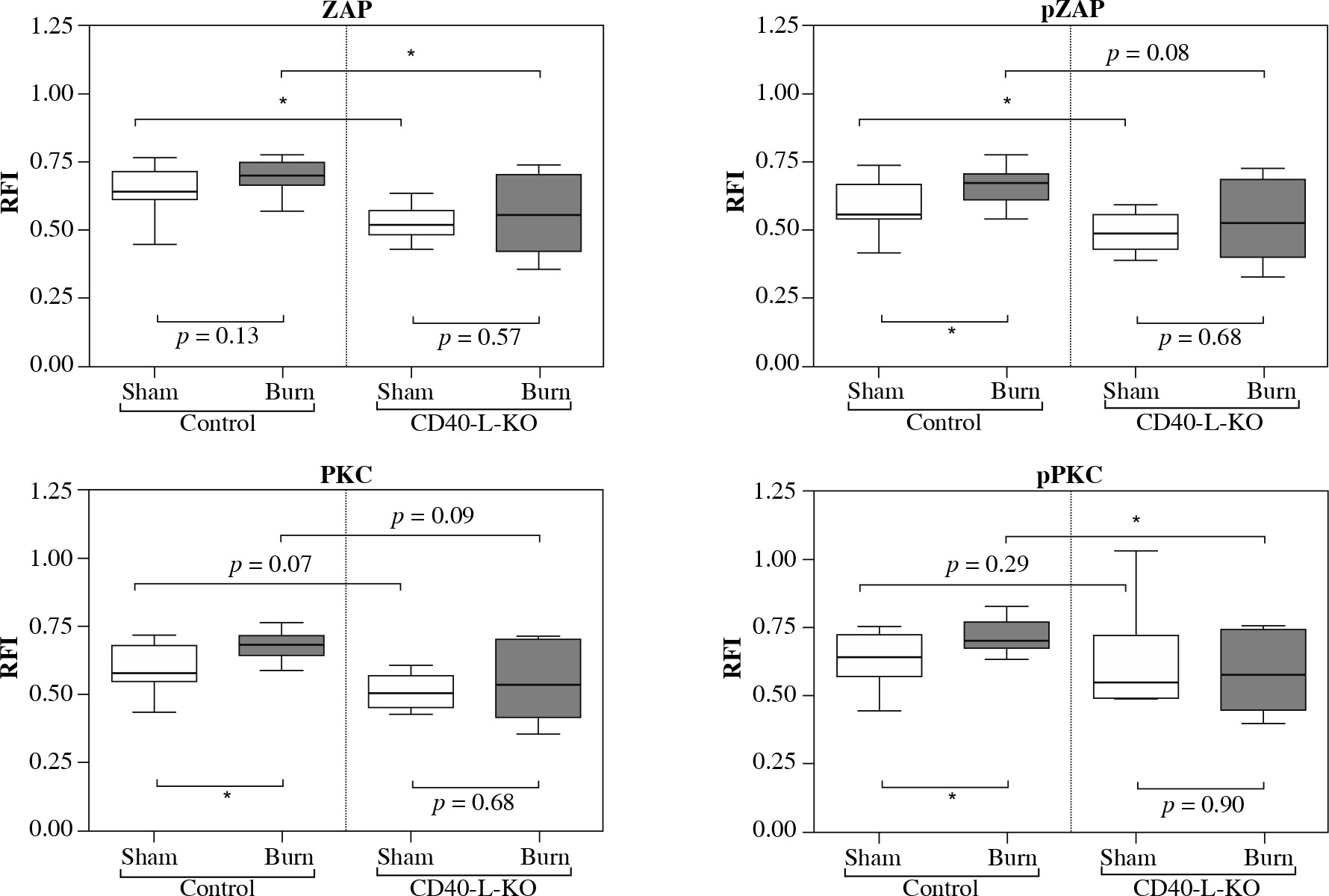
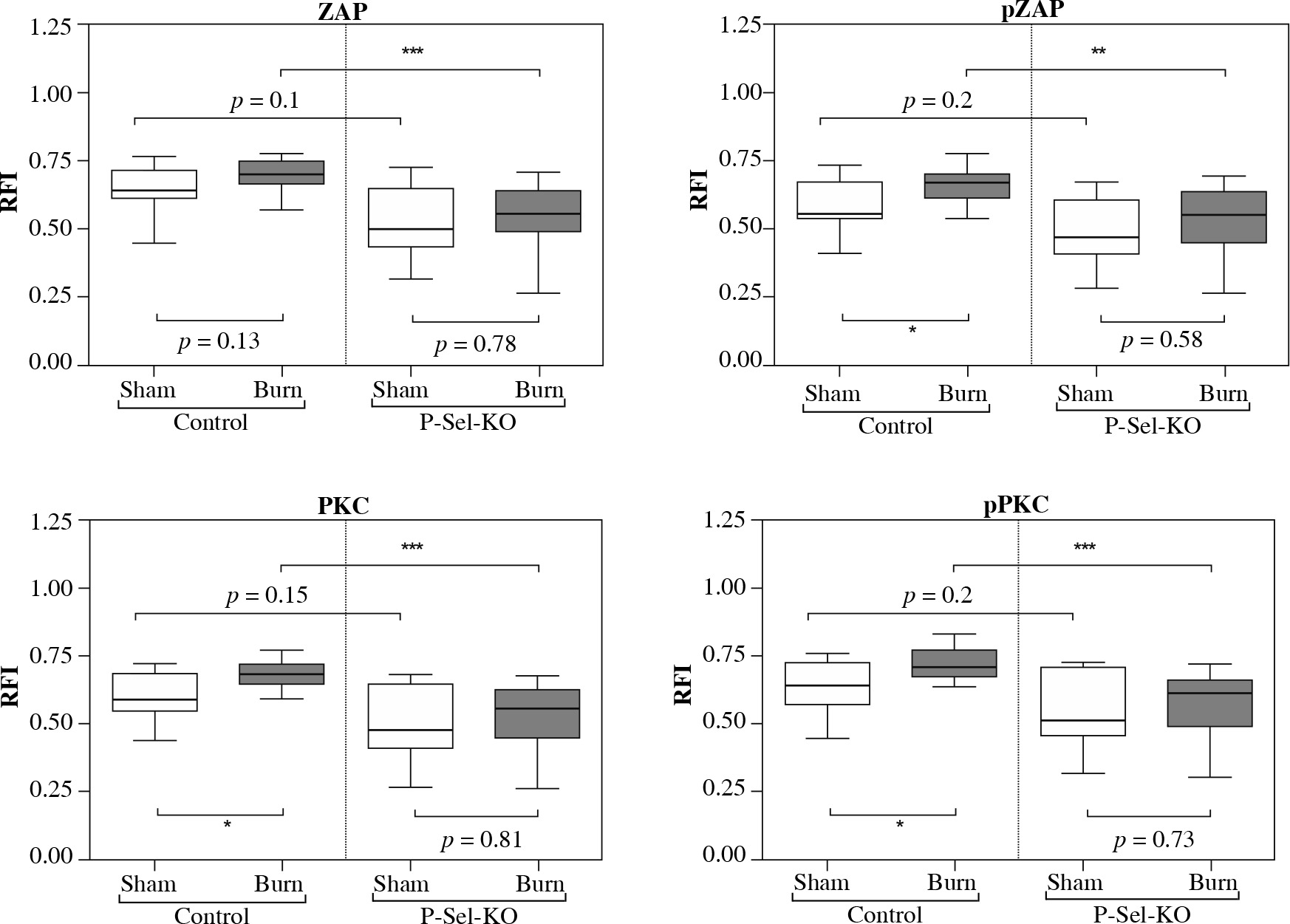
Fig. 3
Baseline activity, posttraumatic activation and posttraumatic activity in lymph node based CD4+ Tregs are lower in P-selectin-KO animals than in WT animals. The phospho-flow cytometric signal at baseline of lymph node based CD4+ Tregs is lower in P-selectin-KO animals than in WT animals. Trauma leads to a significant posttraumatic increase of the phospho-flow cytometric signal of CD4+ Tregs in WT animals (significant for pZAP, PKC, pPKC). In contrast, the posttraumatic phospho-flow cytometric signal is diminished in P-selectin-KO animals and settles at a significantly lower level of activity compared to WT animals. Boxes: Median ± Q1 and Q3, whiskers: min. and max value of each data set, n = 7-15 per group, *p < 0.05, **p < 0.01, ***p < 0.001
CD40L-KO deficiency results in lower posttraumatic activation of lymph node derived CD4+ Tregs
Comparing the results of fluorescence intensity measurement of CD4+ Tregs in the draining lymph nodes between a wild-type control and CD40L gene knockout (Fig. 2), we chose baseline activity (= CD4+ Treg activity of sham-treated animals) and posttraumatic activation as functional parameters of interest in the CD4+ Treg cells. We observed the baseline activity of lymph node derived CD4+ Tregs to be consistently lower for all tested signaling molecules in CD40L-KO-deficient compared to wild type animals with △ZAP-70: 0.12 (p < 0.05), △pZAP-70: 0.07 (p < 0.05), △PKC-δ: 0.07 (p = 0.07), △pPKC-δ: 0.09 (p = 0.29). In line with previous findings [10, 16, 17], we observed that trauma led to biologically significant posttraumatic activation of CD4+ Tregs in WT animals one hour after trauma. Of interest, here we were able to show for the first time that the posttraumatic activation is dampened in CD40L-KO animals with △ZAP-70: 0.04 (p = 0.57), △pZAP-70: 0.04 (p = 0.68), △PKC-δ: 0.03 (p = 0.68), △pPKC-δ: 0.02 (p = 0.9) when comparing the sham-treated group versus the burn-intervention group. Furthermore, the posttraumatic activity of CD4+ Tregs in CD40L deficiency settles at a significantly lower level of activity compared to WT animals, when disrupting CD40L-dependent signaling cascades, with △ZAP-70: 0.14 (p < 0.05), △pZAP-70: 0.15 (p = 0.08), △PKC-δ: 0.14 (p = 0.09), △pPKC-δ: 0.13 (p < 0.05).
Disruption of P-selectin-mediated signaling dampens the posttraumatic activation of lymph node based CD4+ Tregs
In a second set of experiments, we compared the differences in CD4+ Treg activation between a wild-type control and knockout animals deficient in P-selectin (Fig. 3).
We observed the fluorescence intensity derived baseline activity of lymph node derived CD4+ Tregs to be consistently lower in P-selectin-KO animals than in WT animals for all tested signaling molecules, with △ZAP-70: 0.14 (p = 0.1), △pZAP-70: 0.09 (p = 0.2), △PKC-δ: 0.11 (p = 0.15), △pPKC-δ: 0.13 (p = 0.02). In contrast to the findings in WT animals, the posttraumatic activation is diminished in P-selectin-KO animals, comparing the sham-treated group with the burn-intervention group, resulting in △ZAP-70: 0.06 (p = 0.78), △pZAP-70: 0.08 (p = 0.58), △PKC-δ: 0.08 (p = 0.81), △pPKC-δ: 0.10 (p = 0.73). Furthermore, the posttraumatic activity of CD4+ Tregs in P-selectin deficiency settles at a significantly lower level of activity compared to WT animals when disrupting P-selectin-dependent signal cascades, with △ZAP-70: 0.20 (p < 0.001), △pZAP-70: 0.21 (p < 0.001), △PKC-δ: 0.21 (p < 0.001), △pPKC-δ: 0.20 (p < 0.001).
Alteration in hemostatic function in CD40L- and P-selectin deficiency
We used rotational thromboelastometry (ROTEM delta) to examine the effects of trauma on platelet activation and function in WT animals as well as CD40L and P-selectin KO animals respectively (Fig. 4).
Fig. 4
Alteration in hemostatic function in CD40L- and P-selectin deficiency. As of rotational thromboelastometry (ROTEM) measurements, the clotting time (CT) in non-traumatized animals is prolonged in CD40L-KO- and P-selectin-KO animals compared to WT animals. When traumatized, the CT is not significantly altered compared to the non-traumatized state, either in the control or the deficiency group. The maximum clot firmness (MCF) tends to be reduced in CD40L-KO and is significantly reduced in P-selectin-KO compared to WT animals. Upon trauma, the MCF does not significantly change compared to the non-traumatized level in WT animals as well as in non-traumatized CD40L- and P-selectin-deficient animals. The speed of clot formation, measured by alpha angle and clot formation time (CFT), is reduced in non-traumatized CD40L-KO-mice compared to non-traumatized WT animals while no significant changes in CFT and alpha-angle are observed in P-selectin-deficiency. Boxes: Median ± Q1 and Q3, whiskers: min. and max value of each data set, n = 7-15 per group, *p < 0.05, **p < 0.01, ***p < 0.001
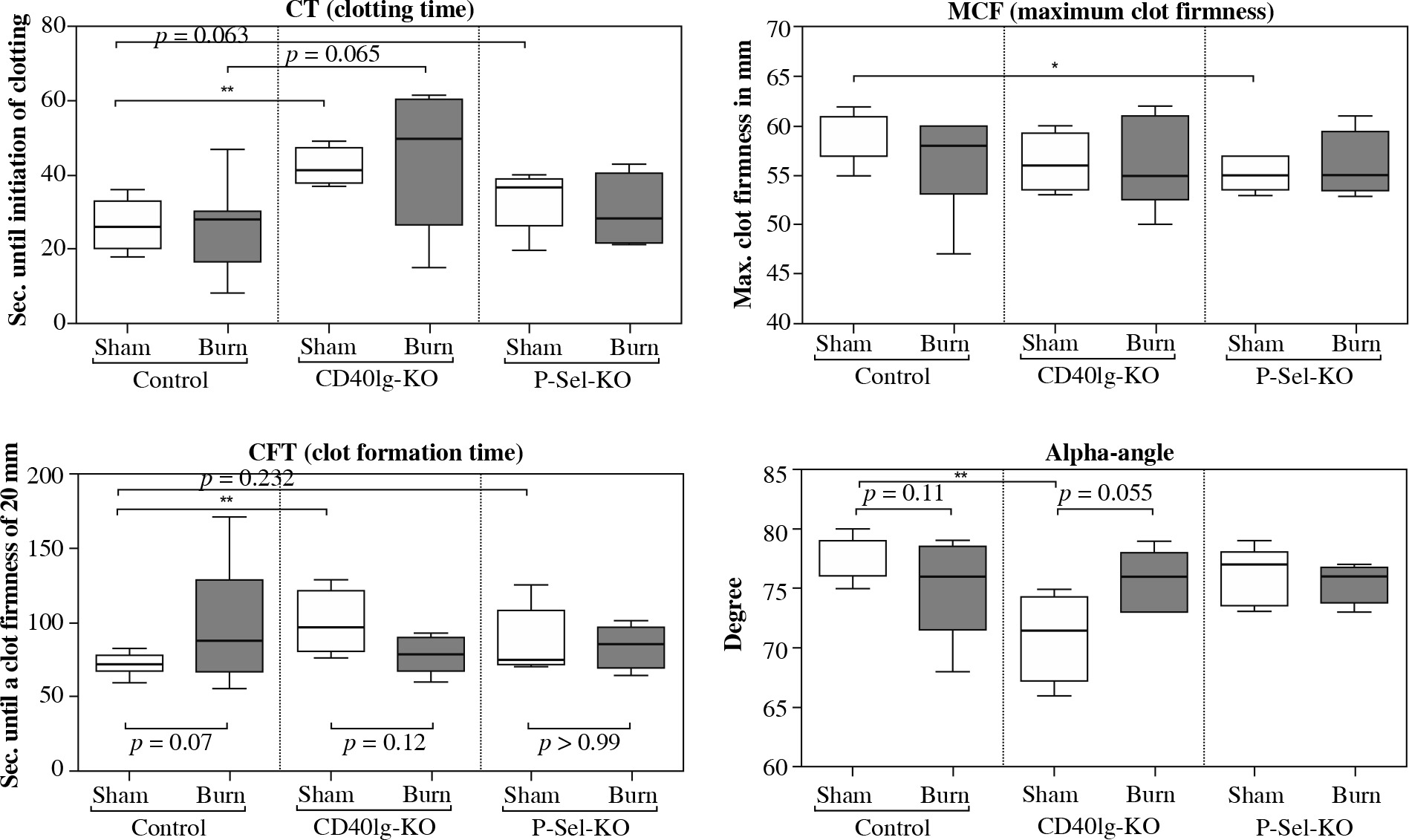
Our results suggest an influence of CD40L/P-selectin-dependent pathways on platelet activation and function. The time of initiation of hemostasis – the CT – in non-traumatized animals is prolonged in P-selectin-KO and significantly prolonged for CD40L-KO animals compared to WT animals. When traumatized, the CT is not significantly altered compared to the non-traumatized state, either in WT animals or in CD40L-KO- or P-selectin-KO animals. The MCF is significantly reduced in P-selectin-deficient animals compared to WT animals, while for CD40L-deficiency, no such effect is observable close to statistical significance. Upon trauma, the MCF remains largely stable compared to the non-traumatized level in WT animals as well as CD40L- and P-selectin deficiency. The speed of clot formation (CFT and alpha angle) is significantly reduced in CD40L-deficient compared to WT animals, while P-selectin deficiency does not significantly change the CFT and alpha angle measurements. Upon trauma in wild type animals, the velocity of clot formation tends to be reduced. Contrary to that, in CD40L-deficient animals, the data show a trend towards faster clot formation posttraumatically. P-selectin deficiency does not seem to affect the velocity of clot formation in the event of trauma.
In summary, CD40L deficiency delays initiation of clotting and prolongs platelet-associated clot formation as observed via rotational thromboelastometry. P-selectin deficiency delays initiation of clotting and impairs clot firmness. Posttraumatically, deficiency in neither of the molecules imitates the results after trauma when depleting CD4+ Tregs in previous studies, which indicated hypocoagulability [16].
Discussion
CD4+ Tregs are considered key players in the orchestrated immunologic response of the host to trauma, controlling the adaptive immune response by suppressing T cell activation [34]. The pathways of CD4+ Treg activation are subject to intensive research. Previous experiments from the co-authors assessing CD4+ Treg activation early following trauma show activation of TCR signaling pathways following trauma [10] and the engagement of CD4+ Tregs in a reciprocally activating interaction with platelets [16]. In order to identify potential mechanisms of the posttraumatic interaction between CD4+ Tregs and platelets, we investigated pathways of cell-cell contact signaling in this screening study and found that CD40L and P-selectin affect the posttraumatic activation of CD4+ Tregs and platelets.
To detect early CD4+ Treg activation, we used phospho-flow cytometry for intracellular CD4+ Treg signaling molecules as previously established [10, 30] due to the following advantages. The high-throughput capacity of flow cytometry enables powerful screening of different cells, specific staining and gating techniques enable the accurate identification of the cell types, and the results are quantifiable and comparable [35]. With phospho-flow cytometry, we measured the expression and phosphorylation of the intracellular T cell activation molecules ZAP-70 and PKC-δ, as they allow for detection of both TCR-dependent (ZAP-70, PKC-δ) and TCR-independent (PKC-δ) pathways of activation and are relatively T cell specific [30]. To assess the behavior of platelets in this reciprocal interaction, we employed rotational thromboelastometry, a method to analyze platelet activation and function [36].
Having gained insight on paracrine interactions via TNFR2-, TLR4- and IL-10-dependent pathways in previous experiments [17], we assessed the potential influence of direct cell-cell contact signaling via a costimulatory (CD40L) and an adhesion molecule (P-selectin) in this screening study. Both P-selectin and CD40L are considered important in CD4+ Treg development and function [22, 25, 29], but previous studies assessed that in knockouts, CD4+ Tregs are – though in different quantities – still generated and functional [22, 29, 32, 37]. Analyzing cell-counts and percentages, we could quantitatively confirm these findings.
We employed the burn injury model, which is known to reliably and consistently stimulate CD4+ Tregs as a part of the hosts immune response [9, 10]. While yielding a relatively low injury-induced mortality (< 5%), it validly simulates the clinical immunologic condition of severely injured patients [38].
Cellular phenotype in CD40L and P-selectin-deficiency
In line with the findings in the present study, previous studies in rodents have shown that both antibody-mediated blockage of CD40L [21] as well as CD40L-deficiency [22, 37] result in a reduction of the absolute number of CD4+ Tregs, with the overall ratio of T cell subpopulations however remaining physiologic [32]. Conversely, again in line with the findings in this study, Kohm et al. demonstrated a 60-80% increase in CD4+ Tregs compared to wild-type animals. Combined, these finding support the suggested role of CD40L and P-selectin in the complex development and overall homeostasis of the CD4+ Treg population [29, 39].
CD40L deficiency results in lower activation of CD4+ Tregs
Current theories about posttraumatic activation CD4+ Treg based on the “danger” theory proposed by Matzinger et al. hypothesize that alarmins may activate CD4+ Tregs through indirect activation via presentation for the TCR on the APCs’ MHC-peptide complex [34, 40]. For TCR-dependent T cell activation, costimulatory signaling is necessary for full scale effect [41]. Therefore, we investigated CD40L, an activating costimulatory molecule on the CD4+ Treg-surface from the TNF superfamily [41, 42], for a possible role in the posttraumatic CD4+ Treg-activation. Platelets are bearing CD40 – the receptor of CD40L [19, 24] – and a reciprocal interaction of T cells and platelets via CD40-CD40L has already been observed in non-traumatic settings [18, 19].
Indeed, we could observe changes in posttraumatic CD4+ Treg and platelet function upon CD40L deficiency. The activation parameters of lymph node based CD4+ Tregs are significantly dampened in CD40L deficiency. As Bergmann et al. observed similar changes upon depletion of platelets [16], significant signaling of the CD4+ Treg-platelet-interaction may potentially occur via CD40L-dependent pathways.
Different molecular mechanisms, by which CD4+ Tregs can be activated via CD40L from platelets, could explain our findings. Realizing its costimulatory potential, CD40-CD40L ligation can – via TRAF-dependent initiation of MAPK, PLCγ, PI3K and NFκB signaling cascades or via JAK3-mediated STAT5-phosphorylation – lead to CD4+ Treg-cell activation and immunologic effector functions [42]. Downstream pathways of a CD40L ligation to APC-expressed CD40 can include subsequent modulation of the costimulatory molecule B7, in turn causing enhanced CD4+ Treg activation via B7-CD28 interaction [43]. This mechanism has been investigated in studies that show that platelets express B7 and activate T cells via CD28-B7-dependent pathways [44]. Alternatively, via a paracrine mechanism, CD4+ Treg-CD40L could stimulate platelets to release granules stores rich in RANTES (regulated and normal T cell expressed and secreted), which instantly mediates further T cell recruitment, creating a reciprocally activating amplifying feedback loop [19]. Further studies to clarify these potential activation mechanisms are required.
Having observed an effect on early CD4+ Treg activation upon disruption of CD40L-signaling, CD4+ Treg functionality remains subject to intensified research. Studies in rodents have shown that antibody-blockade of CD40L – in line with our findings and other studies in CD40L gene-deficiency [22, 37] – cause a drop in the absolute number of CD4+ Tregs [21]. The suppressive capacity of CD4+ Tregs seems to remain intact upon CD40L blockade and deficiency [22] as well as extended costimulation-blockade (including CD28) [23]. Interestingly, CD4+ Tregs seem to be less dependent on CD40– and CD28-costimulation for their effector function compared to CD4+non-Tregs [23]. This effect is to be considered when concepting increasingly translational studies that involve CD40L-CD40 as a target molecule.
Our finding of posttraumatic cell-cell-contact-dependent activation of CD4+ Tregs via CD40L fits well with latest concepts of T cell activation and provides new insights of the mechanisms of the posttraumatic activation of CD4+-Treg. As soluble CD40L is considered a predictive proinflammatory marker posttraumatically [45], our findings add to the understanding of the posttraumatic role of CD40L by observing a role in a possibly protective anti-inflammatory process.
CD40L deficiency results in altered hemostatic parameters
To our knowledge, this is the first study to investigate dynamic hemostatic parameters via ROTEM in CD40L-deficiency, demonstrating that the initiation of hemostasis (CT) is significantly prolonged and the speed of clot formation (CFT and alpha angle) is significantly reduced in CD40L-deficiency. These results can be interpreted along the understanding of membrane-bound CD40L as a platelet co-activator [46]. More specifically, CD40L can activate platelets directly via ligation of CD40 and GPIIb/IIIa and indirectly via induction of TF on monocytes [47]. Consecutively, CD40L-ligation of GPIIb/IIIa is important for platelet aggregation, stability of arterial thrombi, fibrinogen binding, thrombin-mediated activation of platelets and the induction of a self-activating amplification loop by upregulating CD40L surface exposure on platelets [46, 47], while an upregulated expression of TF triggers the extrinsic coagulation cascade [48].
In summary, we show that latency and speed of clot formation are prolonged in CD40L deficiency, but no significant posttraumatic changes could be identified compared to WT animals. The trend towards hypocoagulation early after trauma, when interrupting CD4+ Treg-platelets interaction [16], does not seem to be attributable to the CD40L deficiency. Yet our results show CD40L to be involved in adequate hemostasis considered protective for traumatized patients.
Disruption of P-selectin-mediated signaling dampens the posttraumatic activation of lymph node based CD4+ Tregs
An initial step in a T cell-mediated inflammatory response is the recruitment of T cells into responding lymph nodes or inflamed tissue via the adhesion molecules P-selectin and PSGL-1 [49]. A specific platelet-CD4+ T cell interaction via direct cell-cell contact in a P-selectin-dependent fashion could be documented via intravital microscopy in liver ischemia reperfusion [18]. Based on the significantly elevated expression of P-selectin in CD4+ Tregs compared to CD4+ non-Tregs [29], we investigated its possible role in direct cell contact signaling of CD4+ Tregs and platelets.
Indeed, in P-selectin deficiency, all activation parameters of CD4+ Tregs are significantly lowered. As Bergmann et al. observed similar changes when completely depleting platelets [16], the results of this screening study suggest that significant signaling of the CD4+ Treg-platelet interaction may potentially occur via P-selectin-dependent pathways.
These results are supported by the molecular understanding of P-selectin-dependent signaling. Posttraumatically stimulated platelets upregulate P-selectin expression, leading to their initial binding to DAMP-activated leukocytes expressing PSGL-1 [50] and the initiation of downstream signaling pathways. Firstly, the intracellular signaling of PSGL-1 via the adapter protein moesin, known to regulate T cell-activation through Syk-kinases, may play a role in CD4+ Treg activation [51]. Secondly, indirect P-selectin-associated CD4+ Treg activation may be mediated via RANTES that can instantly mediate T cell recruitment [19]. Thirdly, firm adhesion of platelets and CD4+ Tregs can facilitate the T cell activating capacity of costimulatory molecules such as CD40L. By ensuring local proximity via P-selectin-dependent adhesion, the known activation via paracrine interactions between CD4+ Tregs and platelets, e.g. via TNF-α or TLR4, can exhibit their full stimulatory potential [17]. The potential role of P-selectin-PSGL-1 in early posttraumatic activation of CD4+ Tregs observed in this study expands the understanding of P-selectin to the relevant role in anti-inflammation. This has been suggested previously in the adaptive immune system, as PSGL-1 has been observed to negatively regulate T cell immune responses [52]. Furthermore, an inflammatory immunologic phenotype caused by an imbalance between proinflammatory effector cells and regulatory cells has been associated with PSGL-1 deficiency [27]. The role of PSGL-1 as an immune checkpoint regulator has been discussed, when observing that PSGL-1 deficiency is associated with increased levels of proinflammatory TNF and interferon γ while expressing decreased levels of inhibitory receptors such as programmed cell death-1 (PD-1) in T effector cells [53].
Indeed, the role of P-selectin-dependent signaling is not limited to the early CD4-Treg activation observed in this study, but P-selectin is further considered to be a crucial molecule for effective CD4+ Treg development. It is known that in P-selectin deficiency, CD4+ Tregs are significantly less effective in suppressing CD4+non-Treg proliferation, while not directly influencing CD4+ Treg effector function [29]. Investigating this regulatory capacity of P-selectin-PSGL-1, it was observed that CD4+ Treg development is induced by dendritic cells that express a tolerogenic receptor portfolio upon PSGL-1 signaling and induce differentiation of naive CD4+ lymphocytes into CD4+ Tregs [54]. Considering this ongoing discussion, our findings add to the understanding of the CD4+ Treg-associated posttraumatic role of P-selectin, suggesting a potential role of the molecule in protective anti-inflammatory processes.
Disruption of P-selectin-mediated signaling influences hemostatic parameters
In our ROTEM measurements in P-selectin deficiency, the clotting time tends to be prolonged and maximum clot firmness is significantly reduced, while the posttraumatic hemostatic function is not significantly altered compared to WT animals. These results reflect the current understanding of the activating role of P-selectin in hemostasis. Platelet P-selectin was observed to have procoagulatory action by inducing TF [55] and mediating fibrin incorporation and leukocyte accumulation in the thrombus, so that in P-selectin deficiency, a prolonged bleeding time and abnormal thrombus formation can be observed [56]. Montoro-Garcia et al. also attributed procoagulatory effects to P-selectin-rich sPMP derived from activated platelets [57]. These intrinsic procoagulatory features of P-selectin may reflect the reason for the reduced coagulatory parameters in our thromboelastometry measurements. As no significant posttraumatic changes could be identified in P-selectin deficiency compared to WT animals, the trend towards hypocoagulation early after trauma, when interrupting CD4+ Treg-platelet interaction [16], does not seem to be attributable to disruption of P-selectin pathways. Yet, the results dynamically demonstrate P-selectin-dependent pathways to be involved in adequate hemostasis considered protective for traumatized patients.
In the aftermath of trauma, CD4+ Tregs have been shown to regulate immunity by suppressing CD4+ T effector cell function and thereby counter systemic hyperinflammation [8]. The CD4+ Treg function as well as the expression of interaction molecules has been shown to be increased posttraumatically [9]. These changes in CD4+ Treg biology have also been observed in human studies investigating hyperinflammation [58, 59]. In further understanding the complex interactions leading to immunologic imbalance following trauma, our results provide new insights into the activation of protective posttraumatic mechanisms.
While this screening study has provided interesting findings, further investigations including flow cytometry to determine molecule expression and cell culture methods to investigate cell-cell contact are required.
Conclusions
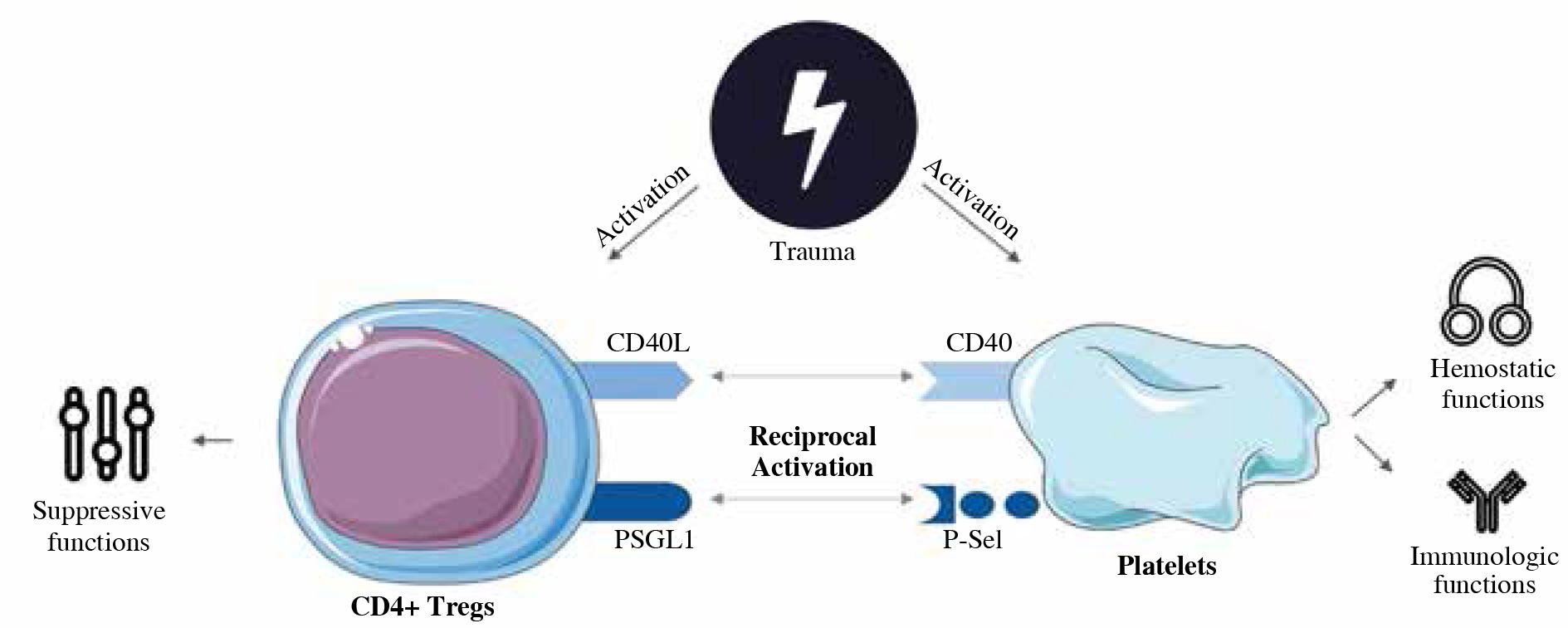
In conclusion, we described the role of direct cell-cell contact pathways in the interaction between CD4+ Tregs and platelets early after trauma for the first time (Fig. 5) [60]. Though immune perturbation is a relevant clinical problem in management of traumatized patients, the current concepts of immunosurveillance remain insufficient and lack the clear mechanistic understanding needed to derive actionable insight. Our study provides an insight into the role of direct cell-cell-contact-dependent mechanisms in the activation of posttraumatic immunologic players enabling a better understanding of parameters of interest in posttraumatic immunosurveillance. This marks a further step towards improved monitoring and tailored interventions that yield the potential for a more effective treatment of the complex pathological posttraumatic immune process. Furthermore, as an adequate hemostatic balance is considered crucial for the patients’ posttraumatic clinical outcome, our study provides a mechanistic understanding of the role of P-selectin and CD40L in platelet-dependent posttraumatic hemostasis. The impact of these pathways on hemostasis has to be considered when employing CD40L- and P-selectin-based immunomodulatory interventional strategies.
Fig. 5
Schematic concept of the role of direct cell-cell contact via CD40L and P-selectin in early posttraumatic interaction of CD4+ Tregs and platelets. Under normal circumstances, CD4+ Tregs ensure immunologic balance and tolerance with their suppressive capabilities [60]. Upon trauma, CD4+ Tregs are activated and exhibit enhanced suppressive capabilities to counter posttraumatic hyperinflammation [7]. If CD40L- or P-selectin-dependent pathways are interrupted, posttraumatic activation is diminished (Figs. 2 and 3), suggesting that CD40L and P-selectin play a relevant role for CD4+ Treg activation and therefore a satisfactory posttraumatic immunosuppressive response to counter posttraumatic hyperinflammation. Furthermore, hemostatic functions seem to be CD40L- and P-selectin- pathway dependent to a certain degree, as deficiency of the molecules delays initiation of clotting and additionally CD40L deficiency prolongs platelet-associated clot formation and P-selectin-deficiency impairs clot firmness. Posttraumatically, CD4+ Tregs do not seem to influence platelet function via CD40L- or P-selectin-mediated direct cell-cell contact