
Introduction
Dysmorphic syndromes are defined as specific sets of symptoms that result from common pathogenic mechanisms. These include both visible differences in facial features or body proportions and more serious congenital defects of organs, which arise in the earliest stages of embryonic development [1, 2]. In clinical practice, this term refers to the comprehensive evaluation of body shape and individual anatomical features that deviate from the established norms for a given population in terms of ethnicity, age, and gender. It is imperative to consider the patient’s family background, with particular attention to the physical characteristics of the parents, to ensure that erroneous conclusions arising from an overreliance on the analysis of the patient’s face or head alone are avoided. The present article discusses dysmorphic syndrome in the context of excessive growth syndrome in patients, which is characterized by excessive growth in children, determined by measuring body length using an Epstein bench or infant stadiometer. Excessive growth is defined as body length/height above the 97th percentile [3, 4]. This paper discusses syndromes characterized by excessive growth, which are caused by chromosomal abnormalities and methylation disorders which disrupt the proper regulation of imprinted genes. Such conditions are rare and frequently complex, arising from abnormalities in the structure or number of chromosomes. These abnormalities lead to disturbances in the regulation of the body’s growth processes. Such anomalies may encompass both substantial deletions or duplications of chromosome fragments, as well as more nuanced alterations, such as microdeletions or microduplications, impacting the expression of genes that regulate cell development and proliferation [5–7]. The objective of this paper is to discuss the molecular mechanisms, characteristic clinical phenotypes, and diagnostic and differential possibilities of these syndromes in order to facilitate their recognition in clinical practice.
Methodology
A comprehensive literature search was conducted using Google Scholar, PubMed, Web of Science, Orphanet (ORPHA), Online Mendelian Inheritance in Man (OMIM), and the National Institutes of Health database. The following search phrases were applied to ensure a thorough examination of the topic:
causes of the disease,
clinical presentation of the disease,
diagnosis of the disease,
treatment of the disease.
The selection process was guided by specific inclusion criteria to ensure the relevance, quality, and reliability of the data. We prioritized studies that:
were as recent as possible, to reflect the latest scientific findings,
included case reports, which offer detailed descriptions of clinical manifestations and treatment outcomes,
were original, peer-reviewed, and published research articles to ensure scientific rigor,
had full-text availability, allowing for comprehensive analysis,
were written entirely in English, to maintain consistency and facilitate interpretation.
Applying these criteria, a total of 50,385 studies were identified, of which 36 were included in the final analysis.
Results
Fragile X syndrome
Fragile X syndrome (FXS) [OMIM: 300624, ORPHA: 908] is caused by a mutation in the FMR1 gene, which is located on the X chromosome in the Xq27.3 region [8, 9]. The disorder has been defined as a neurodevelopmental and neurodegenerative disease, reflecting its complex effects on nervous system function. Mutation in the FMR1 gene leads to abnormal expression of the fragile X mental retardation protein (FMRP), which is crucial for normal development and function of synapses, resulting in impaired brain plasticity and difficulties in cognitive and behavioural processes [9]. The estimated birth prevalence of FXS is approximately 1–5 per 10,000 live births, occurring in about 1 in 7,000 males and 1 in 11,000 females [10].
The diagnosis of an FMR1 disorder is established through the use of specialized molecular genetic testing to detect cytosine–guanine–guanine (CGG) trinucleotide repeat expansion in the 5′ untranslated region of FMR1 with abnormal gene methylation for most alleles with > 200 repeats. Typically, a definite diagnosis of FXS requires the presence of a full-mutation repeat size (> 200 CGG repeats), while the diagnosis of fragile X-associated tremor/ataxia syndrome or fragile X-associated primary ovarian insufficiency is associated with a premutation-sized repeat (55–200 CGG repeats).
The syndrome is characterized by moderately to severely impaired intellectual development and prominent facial features (e.g. a long face, large ears and a prominent jaw) [8]. A number of characteristic morphological features have been observed among men diagnosed with FXS. The most prominent symptom is macroorchidism, or enlarged testes, which is one of the key elements of the clinical picture. These patients often present with hypoplasia of the midface, which contributes to a peculiar appearance with delicate but recognizable features, including thick lips, a markedly enlarged jaw and a prominent chin. High-arched palates and underdeveloped ears are additional characteristic features of those affected by FXS (Figure 1). Furthermore, patients with the syndrome show varying degrees of intellectual disability [11], alongside delays in language and motor development that often lead to difficulties in communication and in performing daily activities [12)]. Characteristic behavioural and neuropsychiatric features include mood lability, anxiety, impulsivity, aggressiveness and hyperactivity [13, 14].
Figure 1
Dysmorphic features of Fragile X syndrome (the authors’ own collection; photos published with the consent of guardians)
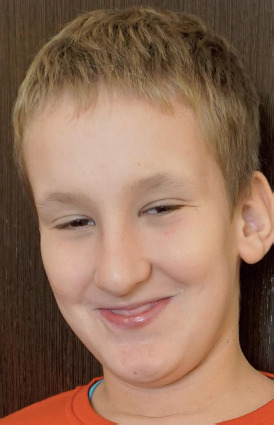
The diagnosis is made based on the clinical picture and ancillary investigations. Among these investigations, magnetic resonance imaging (MRI) is distinguished by its ability to show periventricular heterotopia [15] and significantly reduced cortical lobes, the size of the posterior cerebellum, the amygdala and the superior temporal lobe, and an increase in the size of the caudate nucleus. The reduced expression of FMRP, along with the presence of cognitive deficits, was found to be attributable to the small posterior cerebellar area and the enlarged caudate nucleus [16]. In the course of conducting diagnostic tests on male subjects, it is important to observe the presence of reduced levels of total cholesterol and low- and high-density lipoproteins [17]. The efficacy and reliability of direct DNA diagnosis should not be overlooked [18–20].
The treatment of FXS involves targeted therapies aimed at reversing the neurobiological abnormalities resulting from FMRP protein deficiency. In addition to approaches targeting modulation of gene expression, drugs such as lovastatin and metformin that affect the regulation of key signalling pathways, including phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin and extracellular signal-regulated kinase/mitogen-activated protein kinase, have shown promise. Modulation of these pathways may lead to improved synapse function, thereby alleviating the neurological and behavioural symptoms observed in FXS patients [21].
Pallister–Killian syndrome
Pallister–Killian syndrome (OMIM: 601803, ORPHA: 884) exhibits mosaicism due to the presence of a supernumerary isochromosome 12p, leading to a total of four copies of the short arm of chromosome 12 rather than the typical two. The estimated birth prevalence is approximately 1 in 20,000 live births [22].
It is a developmental disorder characterized by hypotonia in infancy and early childhood [23], developmental delay, sparse hair [24, 25], unusual skin pigmentation, wide and high forehead, small, upturned nose and protruding upper lips (Figure 2). Pallister–Killian mosaic syndrome may also present with a range of additional features, such as hearing loss, vision impairment, seizures, extra nipples, genital abnormalities, and heart defects. A diverse array of features related to the pulmonary, cardiac, renal, gastrointestinal, genitourinary, ophthalmologic, and auditory systems is also associated with this condition [22, 24, 25].
Figure 2
Dysmorphic features of Pallister–Killian syndrome (the authors’ own collection; photos published with the consent of guardians)
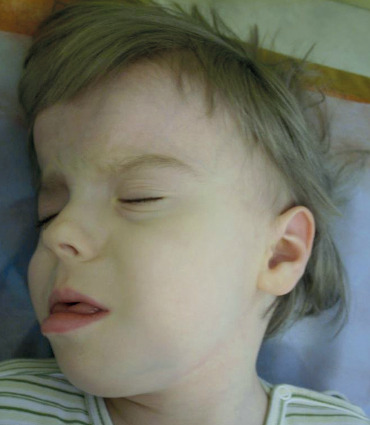
Clinical recognition is essential, as routine blood lymphocyte tests often overlook the presence of an additional chromosome. Fluorescence in situ hybridization was performed on a buccal smear using chromosome 12-specific probes, providing a rapid and non-invasive preliminary diagnosis. However, obtaining a definitive cytogenetic diagnosis may necessitate a skin biopsy and analysis of fibroblast chromosomes due to the variability in sample quality. Typically, the isochromosome is found in 30–100% of fibroblast metaphases. It is crucial to differentiate between mosaic i(12)p and 12p duplication resulting from an unbalanced translocation, as this distinction plays a significant role in genetic counselling [22].
Duplication of chromosome 4 at p16.3
Translocations at 4p16.3 were observed in three families, where deletions led to Pitt–Rogers–Danks syndrome, associated with short stature, while duplications resulted in a distinct overgrowth syndrome. This duplication-related condition was characterized by intellectual disability, coarse facial features, thick hair with bushy eyebrows and synophrys, prominent brow ridges, a square jaw, a small upturned nose, and notably large hands and feet. Affected individuals tended to be taller than average and had an increased head circumference. Although it was proposed that the FGFR3 gene duplication at 4p16 could induce overgrowth due to a dosage effect, this theory did not find support in the available evidence [25].
Chromosome 5p duplication
A full copy of chromosome 5p was identified, along with six other instances. All seven individuals exhibited macrocephaly. A greater birth length was noted in four out of seven cases (one above the 97th percentile, one exceeding the 90th, and two exceeding the 75th) along with a higher birth weight in two out of seven (between the 75th and 90th percentiles). The patients displayed widespread muscle weakness, developmental delays, seizures, hydrocephalus, repeated respiratory infections, and challenges with weight gain. Additional issues consisted of widely spaced eyes, skin folds on the eyelids, a low nasal bridge, an undeveloped midface, a small jaw, abnormally shaped ears, congenital heart issues, airway problems, and clubfoot. The reported cases pinpointed the critical area to proximal 5p (band 5p13) [25].
Chromosome 12p duplication
The genetic disorder chromosome 12p duplication associated with excessive growth syndrome has been documented in 30 clinical cases. The most extensive clinical series documented 16 patients with trisomy 12p, of whom six were over 10 years of age, and demonstrated survival into late childhood and adolescence. The majority of the subjects had normal or elevated birth weight, with a subset exhibiting features of excessive growth and macrocephaly [26].
In the eleven cases under consideration, three exhibited a birth weight in excess of 4000 g, while six exceeded 3600 g. Furthermore, four patients demonstrated a head circumference that was equal to or greater than the 97th percentile. In addition to increased birth weight and macrocephaly, commonly observed features include intellectual disability, decreased muscle tone, short neck, prominent forehead, broad nasal bridge, prominent cheeks, large philtrum, short nose, forward-facing nostrils, widely everted lower lip, and foot abnormalities. The presence of polydactyly and additional nipples has been observed almost exclusively in cases of near-complete duplication of 12p [25].
Duplication of chromosome 12q11-q15
A duplication of chromosome 12q11-q15 was observed in a young child presenting with overgrowth. Birth weight was 6200 g, consistent with extreme macrosomia. By the time the child reached 9 to 10 months old, their height exceeded the 97th percentile. Significant clinical features included low muscle tone, delays in motor skills, an asymmetrical facial structure, sagging eyelids, involuntary eye movements, a prominent palate, large ears, a small pit near the ear, a short neck, an umbilical hernia, tiny hands, a bend in the fifth fingers, and clubfoot [25].
Duplication of chromosome 15q25-qter
The documentation included two patients exhibiting partial duplication of the distal part of chromosome 15. They identified two distinct clinical subtypes based on the location of the duplication: dup(15)(q21-qter) and dup(15)(q25-qter). While there are some shared clinical traits, the second subtype is notably marked by overgrowth, and instances of craniosynostosis have been observed frequently [25].
Deletion involving chromosome 15q12
A patient exhibiting overgrowth had a deletion on chromosome 15(q12). The individual’s birth weight, height, and head circumference were recorded to be above the 97th percentile. Observed clinical characteristics included delayed psychomotor development, a wide forehead, widely spaced eyes, prominent ears, underdeveloped antihelices, and clinodactyly of the fifth finger. The deleted segment in this patient was found to overlap with the Prader–Willi/Angelman region. Additional observations included mild oculocutaneous albinoidism, macrostomia, and predisposition to obesity, with a fat distribution distinct from that typically seen in Prader–Willi syndrome. There were no other signs indicative of either syndrome present. Furthermore, the region of deletion also matched findings from previous reports of Sotos syndrome. Two instances involved deletions on 15q12 or 13, while another patient was noted to have a Robertsonian translocation of 15q;15q [25].
Deletion of chromosome 22q13-qter
A deletion at 22(q13-qter) was detected in an 18-month-old girl who was at the 97th percentile for height, even though her birth weight was 3600 g. Her clinical symptoms included a low-pitched cry, reduced muscle tone, delays in development, fullness of the face, epicanthic folds, a wide nasal bridge, a long philtrum, and a prominent lower lip. In another instance, a child displayed fast growth, loss of hearing, problems with the inner ear, and delayed myelination of the brain. An examination of similar instances highlighted shared characteristics such as decreased muscle tone, developmental delays, excessive growth, drooping eyelids, epicanthic folds, long philtrum, a high-arched palate, ear abnormalities, hearing impairment, and dolichocephaly [25].
Hemihyperplasia
Hemihyperplasia (OMIM: 235000, ORPHA: 2128), with an estimated prevalence of 1 in 13,000 to 1 in 86,000 live births, is characterized by asymmetric overgrowth of one or body regions caused by atypical cell growth [27]. The term “hemihyperplasia” is currently preferred over “hemihypertrophy” because it better reflects increased cell quantity rather than cell size [28]. Isolated hemihyperplasia has been linked to a mosaic paternal uniparental disomy of chromosome 11p15, a region associated with Beckwith–Wiedemann syndrome (BWS). This indicates that isolated hemihyperplasia may be a less severe or localized variant of BWS. Moreover, epigenetic alterations such as hypomethylation of the KCNQ1OT1 (LIT1) gene have been reported in some patients, reinforcing the involvement of dysregulation of imprinted genes [28].
Hemihyperplasia (commonly known as lateral hyperplasia) is often diagnosed at birth and may become more or less noticeable over time. The overgrowth is typically noted at birth or in early childhood [28, 29] and may remain stable or progress over time [28]. Hemihyperplasia can affect specific body segments or specific organs and tissues. Hemihyperplasia can be confined to one side of the body (ipsilateral) or affect opposite sides of the body [28, 30]. Patients often have discrepancies in limb length, asymmetrical facial features or disproportionate growth of certain body parts [28]. Besides physical asymmetry, hemihyperplasia is linked to a higher likelihood of embryonal tumours, especially Wilms tumour, adrenal cell carcinoma, and hepatoblastoma. The total tumour risk in isolated hemihyperplasia is estimated to be 5.9%, with the peak incidence occurring prior to puberty, as shown in Table I [28]. Other associated features may include vascular malformations, lipomas, and skeletal abnormalities such as scoliosis [31].
Table I
Endocrine abnormalities in the described syndromes
The diagnosis of hemihyperplasia is primarily clinical, based on the observation of asymmetric overgrowth. Anthropometric measurements, including limb circumference and length, are essential for quantifying the degree of asymmetry [29]. Radiographic imaging, such as X-rays or MRI, can help assess bony overgrowth and rule out other causes of limb asymmetry, such as vascular or lymphatic malformations [31].
Treatment of hemihyperplasia involves a multidisciplinary approach to address both functional and aesthetic issues. Orthopaedic procedures, such as limb lengthening or epiphysiodesis, may be considered for significant limb length discrepancies [31]. For patients with associated lipomas or vascular malformations, surgical excision or embolization may be necessary, although recurrence is common [32].
Hemihyperplasia-multiple lipomatosis syndrome
Hemihyperplasia-multiple lipomatosis (HHML) syndrome is a syndrome characterized by asymmetric hemihyperplasia, often affecting the extremities, and the presence of multiple, slow-growing, painless lipomas distributed throughout the body [33]. At least six cases of this syndrome have been reported in the literature. Current evidence suggests that the cause of HHML is genetic mutation in the PIK3CA gene, which makes this condition classified as part of PIK3CA-related overgrowth spectrum [34]. Symptoms usually appear in childhood and can vary widely among individuals, including abnormalities of the lymphatic system, enlarged kidneys and vascular malformations [34].
The syndrome is associated with an increased risk of intra-abdominal embryonal malignancies, making regular monitoring essential [34]. Diagnosis is often delayed due to the rarity of the disease [35]. Surgical intervention may be required for lipomas that cause cosmetic or orthopaedic problems, although recurrence is common. Awareness of HHML among healthcare professionals is crucial for timely diagnosis and treatment, as early detection can help mitigate complications and improve patient outcomes [36].
Hemihyperplasia-vascular overgrowth-epidermal nevus syndrome
The hemihyperplasia-vascular overgrowth-epidermal nevus syndrome resembles the HHML syndrome in terms of stable or slowly evolving features but differs in deeply embedded vascular malformations (arteriovenous and lymphatic), less prominent lipomas and the presence of epidermal nevi. There are patches resembling café au lait. The arrangement of the lesions is mosaic [36].
Conclusions
Overgrowth syndromes constitute a rare and heterogeneous group of genetic disorders characterised by excessive growth, distinct dysmorphic features, and variable neurodevelopmental impairment, including intellectual disability, behavioural abnormalities, and delayed motor or language development. The aetiology of these syndromes is frequently intricate, involving chromosomal aberrations such as deletions, duplications, or structural rearrangements, as well as epigenetic dysregulation, particularly abnormal DNA methylation patterns and dynamic mutation. It is evident that imprinted genes and other growth-regulating loci are susceptible to disruption in methylation, a factor that has been demonstrated to contribute to the excessive growth and other phenotypic features that are commonly observed in syndromes.
The diagnosis of this condition is dependent upon a thorough clinical evaluation of the patient’s phenotypic traits, which is then supported by imaging and molecular genetic testing. In select cases, prenatal testing through amniocentesis or chorionic villus sampling may facilitate the identification of these genetic and epigenetic abnormalities prior to birth. However, the majority of diagnoses are typically made postnatally, based on developmental and physical features. The necessity for comprehensive, multidisciplinary care arises from the potential for hormonal imbalances, tumour predisposition, and complex developmental needs in affected individuals.









