
INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory, neurodegenerative, and demyelinating disease of the central nervous system (CNS) affecting approximately 2.8 million people worldwide [1]. It is associated with an important economic burden for society and has a significant impact not only on the quality of patient’s life, but also on their cognitive functions and emotional state [2, 3].
In recent years, new and increasingly effective drugs have been approved for the treatment of MS. The range of highly effective drugs now encompasses the following: ocrelizumab, ofatumumab, natalizumab, alemtuzumab, and mitoxantrone. The following are moderately effective: fingolimod, siponimod, dimethyl fumarate and cladribine. Other drugs include teriflunomide, β-interferons, and glatiramer acetate [4].
The demand for highly effective and well-tolerated drugs for various types of the disease at onset still remains crucial despite new methods of treating MS. Bruton’s tyrosine kinase inhibitors (BTKi) are being developed in the hope of meeting this unmet need.
Currently, MS is considered as an autoimmune disease of the CNS, in the pathogenesis of which B-lymphocytes play a leading role, having several links of influence [5]. Recent studies have proven that B-lymphocytes participate in the presentation of autoantigens to T-lymphocytes, causing their activation and proliferation [5].
Previously, it was believed that T-lymphocytes are the key type of cells mediating inflammatory lesions in MS [6]. It has also been proven that B-lymphocytes secrete pro-inflammatory cytokines such as: lymphotoxin α, tumor necrosis factor, and interleukins (IL-6, IL-10), which can change the inflammatory environment, increasing the activation of T-cells and myeloid cells [7, 8].
B-cells are involved in the formation of ectopic lymphoid follicle-shaped clusters in the CNS responsible for infiltration of the brain and spinal cord [9]. It is possible that neurodegenerative and demyelinating processes can be caused by factors formed in these inflammatory infiltrates [10].
The activation of microglia may also contribute to the development of chronic inflammation, in particular, due to the production of chemokines, cytokines, and reactive oxygen (ROS) and nitrogen (RNS) species [11]. These mechanisms lead to the development of degeneration of neurons, axons, and oligodendrocytes [12]. There are studies proving that microglia activity correlates with the prognosis of the disease and the development of disability in patients with progressive forms of MS [13].
Bruton’s tyrosine kinase (BTK) is an enzyme that plays an important role in the maturation and functioning of B-lymphocytes [14]. This kinase is also involved in the subsequent transmitting signals of chemokine receptors [15] in the release of pro-inflammatory cytokines [16] and integrin activation [17], and also regulates B-cell receptor-mediated processing and antigen presentation [18].
Therefore, BTK plays a leading role in the inflammatory activity of immune cells (i.e., in the activation of B-cells, as well as myeloid cells, such as macrophages and microglia) [19]. BTKi have the ability to penetrate and accumulate in the CNS when the blood-brain barrier (BBB) remains closed [20], since these drugs are low molecular weight agents.
Small molecules have several advantages for biological modulation compared to large molecules, i.e. oral dosing and intracellular action [21, 22]. Therefore, inhibition of BTK is one of the newest approaches in the treatment of MS, as it causes a decrease in the activation and functioning of B-cells, along with the inhibition of microglia activity.
Relapsing remitting (RRMS), secondary progressive (SPMS), and primary progressive (PPMS) forms of MS are the clinical designations that many MS practitioners suppose to be outdated. Relapsing MS (RMS) forms include RRMS and active SPMS, while progressive MS (PMS) encompass non-active SPMS and PPMS [23]. However, this is still the most useful classification of clinical course now being used.
Relapsing forms of MS (RMS) are associated with an active inflammatory mechanism. This is characterized by BBB leakage and focal T-cell and B-cell invasion, which give rise to classic active demyelinating plaques in the white matter. Progressive forms of MS (PMS) are associated with a chronic and slow accumulation of T-cells and B-cells without leakage of the BBB. This type of inflammation creates subpial-demyelinated lesions in the cerebral and cerebellar cortex, causing the slow expansion of pre-existing lesions in the white matter and diffuse chronic inflammation in the normal-appearing white and gray matter [24].
Chronic inflammation is the cause of disability progression in MS. However, to date the management of disability progression remains an unmet need for people with MS, as there are relatively few drugs that have been shown to produce a reduced risk of 3-month confirmed disability progression (CDP) (in particular, siponimod and ocrelizumab). However, in the subgroup with no activity at baseline, according to the phase 3 EXPAND trial of siponimod, a SP1 receptor modulator, demonstrated a limited effect on the 3-month CDP (hazard ratio [HR] = 0.87) [25]. There is evidence that innate immunity (monocytes and macrophages peripherally and B-cells and microglia centrally), which is affected by BTKi, is responsible for persistent neurodegenerative processes and is a significant factor in the progression of disability [26, 27]. BTKi do not lead to chronic B-cell depletion; immune function can be restored in a few days after treatment is stopped, which is important when compared with the stopping of treatment with monoclonal antibodies to CD20, which may take several months [28].
BTKi represent an entirely new class of drugs among disease-modifying therapy (DMT) for MS; several BTKi therapies are undergoing phase III trials (Table 1). BTKi for the treatment of MS are divided into 2 groups depending on the mechanism of inhibition: irreversible (evobrutinib, orelabrutinib, tolebrutinib, remibrutinib) and reversible (fenebrutinib) [29, 30].
Table 1
Clinical trials involving Bruton's tyrosine kinase inhibitors in the treatment of multiple sclerosis
Covalent, irreversible BTK inhibitors bind to the Cys481 residue (cysteine at position 481 of BTK) and block the adenosine triphosphate (ATP) binding pocket, thereby blocking BTK catalytic activity and preventing B-cell receptor signal transduction [32, 33] (Figure I).
Figure I
Schematic diagram of the role of Bruton’s tyrosine kinase in the B-cell signaling. The engagement and aggregation of the B-cell receptor leads to a cascade of changes. This generates a signaling complex consisting of Syk and Lyn. All of this ultimately allows the activation of BTK. Evobrutinib, orelabrutinib, tolebrutinib, and remibrutinib are irreversible BTKi that deactivate BTK by binding to Cys481. Fenebrutinib is a reversible BTKi that deactivates BTK independently of Cys481. Abbreviations: BTK – Bruton’s tyrosine kinase, LYN – Lck/Yes kinase, SYK – spleen tyrosine kinase, Cys481 – cysteine at position 481 of the BTK
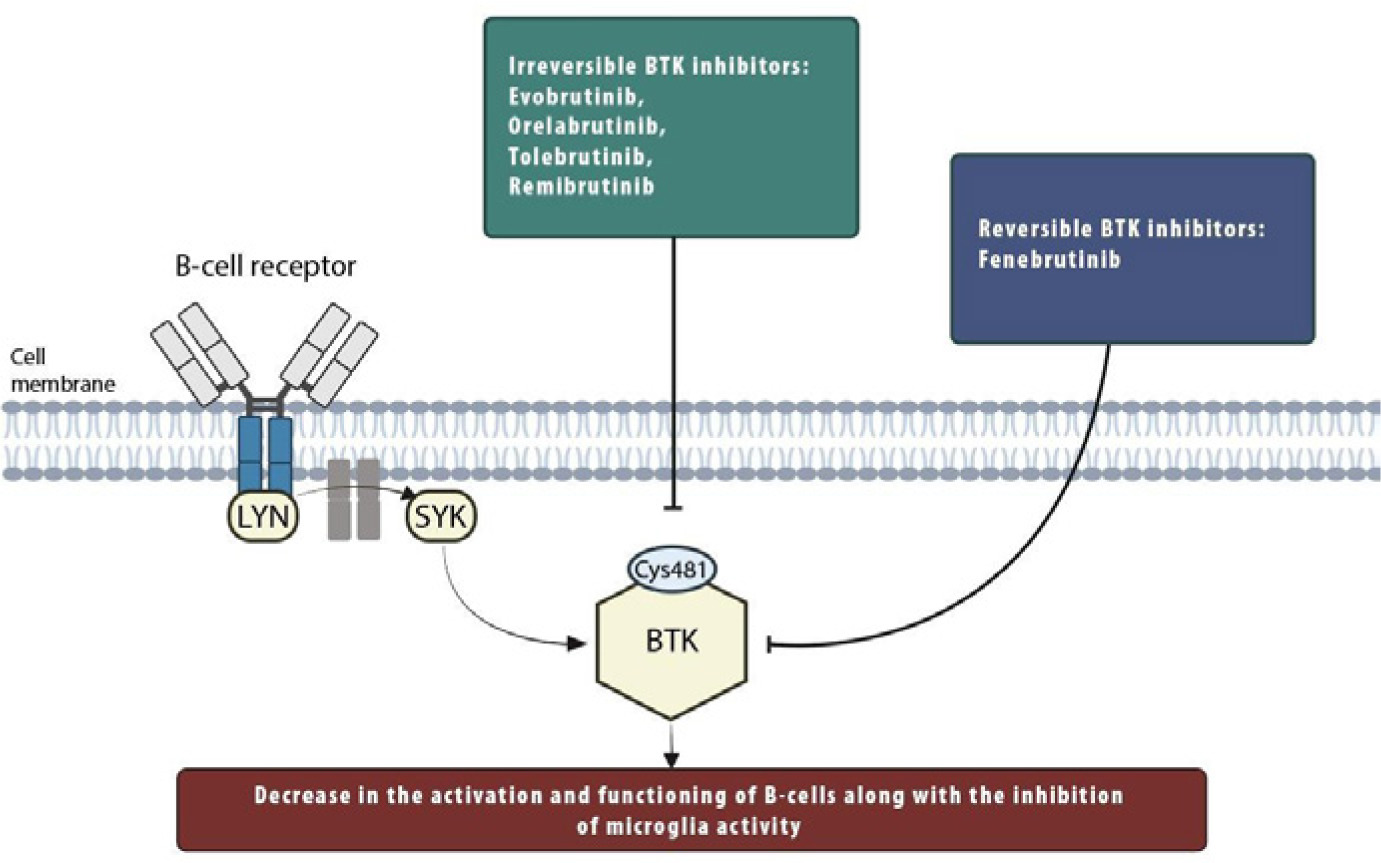
There are several advantages of irreversible, covalent BTKi. Compared to reversible inhibitors, they can potentially demonstrate persistent effects and achieve the full and continuous activity of BTK in target cells, which is necessary for clinical efficacy [30, 31].
Despite the fact that this type of drug has proven itself to be highly effective in the treatment of many diseases, in oncologic indications the problem of acquired resistance to covalent inhibitors has already arisen. Therefore, non-covalent BTKi have been developed to improve the long-term safety of BTKi and pharmacological properties of covalent BTKi, which at the same time are an excellent alternative to solving the problem of acquired resistance, because they do not interact with Cys481, while being extremely selective for BTKi [34, 35].
An advantage of reversible, non-covalent inhibitors is that non-covalent binding is reversible and should have no detrimental effect on the overall effectiveness of the drug, though at the same time the reversible mechanism of inhibition may result in decreased off-target effects that can occur with irreversible covalent inhibitors. Thus, the toxicity and risks associated with the long-term use of a reversible inhibitor may be less than with a covalent inhibitor, which can irreversibly modify off-target proteins [30, 31].
Kinase inhibitors are divided into 5 subtypes, but there are only 4 main ones based on the binding pocket known as the DFG motif (for the amino acid sequence Asp-Phe-Gly) [36, 37]:
Type I: bind competitively to the ATP binding site of active tyrosine kinase.
Type II: bind to inactive kinases in the ATP binding site, and, due to the outward rotation of the DFG motif, regions adjacent to the ATP binding site that would otherwise be inaccessible may be involved.
Type III: bind exclusively to allosteric pockets adjacent to the ATP binding region.
Type IV: bind allosteric sites distant from the ATP binding pocket.
Type V: Drugs that exhibit multiple binding modes [38].
Fenebrutinib is a highly selective, non-covalent reversible BTKi [39]. Fenebrutinib showed favorable pharmacokinetic and pharmacodynamic properties in a single and multiple ascending dose (SAD – single ascending dose, and MAD – multiply ascending dose) in cohort studies of healthy volunteers [40]. Currently, there are 4 ongoing studies evaluating fenebrutinib (Table 1). One of them (NCT05119569) is in a phase II clinical trial, testing drug efficacy in relapsing multiple sclerosis. Participants will be randomized to receive either fenebrutinib or a placebo. Primary outcome measures: the total number of new gadolinium-enhancing T1 lesions observed on MRI scans of the brain. The time frame is 12 weeks.
Two other studies are in phase III clinical trials (NCT04586010, NCT04586023), evaluating drug efficacy regarding disability progression and recurrence rates in adult participants with relapsing multiple sclerosis. Participants will be randomized 1 : 1 to receive fenebrutinib or teriflunomide. Primary outcome measures: annualized relapse rate (ARR). Time frame: minimum of 96 weeks.
The fourth study (NCT04544449) is in a phase III evaluation of fenebrutinib regarding disability progression in adult participants with primary progressive multiple sclerosis. All participants will be randomized 1 : 1 to daily receive oral fenebrutinib (or a placebo) or intravenous (IV) ocrelizumab (or a placebo). Primary outcome measures: time to onset of composite 12-week confirmed disability progression (cCDP12). Time frame: minimum of 120 weeks.
At present, the safety profile of fenebrutinib has been studied in more than 1200 participants regarding several inflammatory diseases; data indicate that the high selectivity of fenebrutinib may limit off-target effects [41]. Bleeding or bruising was observed in 8%, although serious bleeding was rare [42]. Nasopharyngitis, nausea, and headache were observed in > 5% among the conditions reported by the investigators [42].
Irreversible BTKi include evobrutinib, tolebrutinib, orelabrutinib, and remibrutinib.
Evobrutinib is a covalent, irreversible BTKi that is currently undergoing clinical trials for the treatment of autoimmune diseases. The positive effect of this drug in MS is due to its influence on B-cells and myeloid cells, which play a key role in the pathogenesis of MS, and its ability to pass through blood-brain barrier. Three studies are now underway to evaluate the efficacy of evobrutinib (Table 1).
A phase II clinical trial starting in 2017 (NCT02975349) compared evobrutinib at 3 doses (25 mg once daily, 75 mg once daily, and 75 mg twice daily) with a placebo or tecfidera (dimethyl fumarate) in patients with relapsing- remitting multiple sclerosis over 24 weeks to find out about its safety and effectiveness. Primary endpoints constitute the analysis of the number of foci accumulated T1-gadolinium using a magnetic resonance imaging (MRI) scan. Time frame: from 12 to 24 weeks. The results of the study showed that the treatment with evobrutinib resulted in a significant decrease of the neurofilament (NfL) level, a marker of neuroaxonal damage, compared to a placebo over a 2.5-year treatment period [43]. Evobrutinib was generally well tolerated in this clinical trial (NCT02975349) [44]. The most common side effects were urinary tract infections, nasopharyngitis, diarrhea, and elevated alanine aminotransferase [44].
Also, 2 identical phase III studies are currently ongoing for evobrutinib (NCT04338022, NCT04338061). They are investigating the efficacy and safety of evobrutinib (given orally, twice daily) versus teriflunomide (orally, once daily) in patients with relapsing multiple sclerosis. Initial results will assess ARR up to 156 weeks.
Tolebrutinib is also an irreversible covalent BTKi, resulting in a long-term inhibition with direct effects on inflammation in the peripheral and CNS. It has the ability to penetrate the blood-brain barrier, and at the same time powerfully inhibits BTK in microglial cells, preventing the destruction of myelin sheaths [45].
Two identically designed phase III studies are currently ongoing: GEMINI 1 and GEMINI 2 (NCT04410978, NCT04410991) (Table 1). The primary objective of these studies is to evaluate the efficacy of a daily dose of tolebrutinib compared to a daily dose (14 mg) of teriflunomide (Aubagio), calculated by ARR in participants with relapsing forms of MS. Time frame of primary outcome measures: up to approximately 36 months.
Two additional phase III studies, HERCULES and PERSEUS (NCT04411641, NCT04458051), are also ongoing (Table 1). They study drug efficacy compared to placebo in slowing the progression of disability in secondary progressive MS (NCT04411641) and primary progressive MS (NCT04458051). The main outcome for both studies is the time to onset of 6-month confirmed disability progression (CDP). Study duration: up to approximately 48 months.
In June, the FDA partially suspended a phase III clinical trial of tolebrutinib for the treatment of relapsing forms of MS, secondary progressive MS, primary progressive MS, and myasthenia gravis. Registration of new participants in the US, as well as the trial of the drug for the participants enrolling in the trial for less than 60 days, has been terminated.
Accordingly, participants enrolling in studies for more than 60 days will continue to receive treatment. Phase III studies have identified some cases of liver damage. It is significant that the majority of affected participants experienced concomitant complications which predisposed them to liver damage. These lesions were reversible after stopping treatment in all cases [46].
Headache was the most common side effect (in one [3%] of 33 in the group of 5 mg; in three [9%] of 32 in the group of 15 mg, in one [3%] of 33 in the group of 30 mg, in four [13%] of 32 in the group) of 60 mg [47] according to the results of a phase IIb clinical trial of tolebrutinib in 130 patients with relapsing MS and relapsing secondary progressive MS, which was completed on January 2, 2020.
Remibrutinib is a new generation drug that demonstrates high selectivity and efficacy. This means that remibrutinib may potentially have fewer off-target effects [48, 49].
Two identically designed phase III clinical trials are currently ongoing (Table 1), investigating the efficacy and safety of remibrutinib compared to teriflunomide in patients with relapsing multiple sclerosis (NCT05147220, NCT05156281). These studies focused on ARR. Both studies are double-blind, time frames ranging from baseline to 30 months with a subsequent period of up to 5 years.
Although studies of remibrutinib in the treatment of MS have not been completed yet, the administration of remibrutinib was well tolerated in healthy participants and in patients with asymptomatic atopic diathesis at oral doses up to 600 mg. This drug was rapidly absorbed and excreted from the body, and food intake had no noticeable effect on its absorption.
BTK is known to regulate platelet function; consequently, 6 special assessments to identify increased risk of bleeding, including bruising and a coagulation test, were used in the study. As a result, an increased risk of bleeding was not observed at any dose of remibrutinib [50, 51].
Orelabrutinib is a potent second-generation irreversible BTKi targeting both B-cell malignancies and autoimmune diseases, including MS [52].
A randomized, double-blind, placebo-controlled phase II clinical trial (NCT04711148) is currently ongoing in 160 patients aged 18 to 55 with relapsing-remitting MS (Table 1). Patients will be randomly assigned to 1 of 4 treatment groups: placebo, orelabrutinib (low dose), orelabrutinib (medium dose), and orelabrutinib (high dose) at a 1 : 1 : 1 : 1 ratio.
The main objectives of the study are to evaluate the efficacy of orelabrutinib in relation to the cumulative number of new foci in the brain using MRI, T1 accumulated gadolinium in comparison to placebo during 12 weeks of treatment, and to assess safety and tolerability and relapse rates. Time frame of primary outcome measures: up to 120 weeks.
Conclusions
A detailed study of the pathogenesis of multiple sclerosis helps in the development of new drugs. According to recent studies, B-lymphocytes and myeloid cells play a leading role in chronic inflammatory processes of MS. BTKi are drugs that affect precisely these links of pathogenesis.
Currently developed BTKi are low molecular weight agents with an appropriate ability to penetrate the BBB. In this review, clinical studies of the main drugs of this group (evobrutinib, orelabrutinib, tolebrutinib, remibrutinib, and fenebrutinib), their therapeutic benefits and possible side effects were presented. In future, positive results of these studies will be able to significantly enrich the therapy of various forms of MS.






