
INTRODUCTION
Common variable immunodeficiency (CVID) is one of the predominantly antibody deficiencies and the most common symptomatic inborn error of immunity (IEI). It is a primary B cell disorder and specific immunoglobulin (Ig) production is impaired. It is a heterogeneous disease primarily characterized by hypogammaglobulinemia and increased susceptibility to recurrent bacterial and viral infections, including SARS-CoV-2 [1]. CVID also includes a wide range of non-infectious complications and patients present with a wide variety of clinical signs, including autoimmunity, enteropathy, lymphoproliferation, and malignancy [2].
However, in most cases, the genetic background of the disease cannot be identified. Various immunological and genetic defects play a role in the pathogenesis of CVID. B cells are the cells responsible for the humoral immune response, in other words, for specific antibody production. Transcription factors play a role in B cell development and differentiation. One of the genes encoding these proteins is transcription factor 3 (TCF3) [2, 3]. With the TCF3 mutation, there is a deficiency in transcription factor E47 and B cell development is halted. Therefore the humoral response is impaired and recurrent bacterial infections beside non-infectious complications occur [2, 3].
In this report, we present the laboratory results and clinical course of 2 pediatric Turkish cases with de novo heterozygous TCF3 mutations.
CASE REPORTS
Our first case is a 10-year-old Turkish girl who started to have frequent febrile infections, recurrent bronchitis and consequently she was hospitalized many times during this period after the age of 1 year. Intravenous immunoglobulin (IVIG) replacement therapy was started at the age of 2 years. Family history was unremarkable. The patient has been receiving 0.5 g/kg IVIG once a month for 8 years with trimethoprim-sulfamethoxazole prophylaxis. Body weight was 21 kg (SDS: –2.81), height 130 cm (SDS: –1.87), there were bilateral abnormal breath sounds including rales and rhonchi. Immunodeficiency was added to bronchiectasis and growth retardation over time. Hemogram, serological tests, routine biochemistry and flow cytometric evaluation are shown in Table 1. Computed tomography of lung sections of case 1 showed bronchiectatic changes (Figure 1 A). Clinical exome sequencing demonstrated heterozygous missense mutation, c.1750A>G (p.Lys584Glu), a variant of unknown significance, in the TCF3 gene (Figure 1 B). The genetic mutation seems to be compatible with our patient’s clinical table.
Table 1
Immunoglobulin values and specific antibody titers of our cases at diagnosis [14]. Hemograms, lymphocyte subgroup values of our cases at diagnosis [15]
Figure 1
A – Computed tomography of lung sections in case 1. In the lower lobe of the left lung, cystic bronchiectasis areas, peribronchovascular budded tree appearance, and ground glass densities, primarily suggesting nonspecific bronchopneumonia, were observed. B – A heterozygous missense c.1750A>G (p.Lys584Glu / K584E) mutation, variant of unknown significance, is seen in the TCF3 gene. C – A heterozygous missense c.1541C>T (p.Ser514Leu / S514L) mutation, variant of unknown significance, is seen in the TCF3 gene
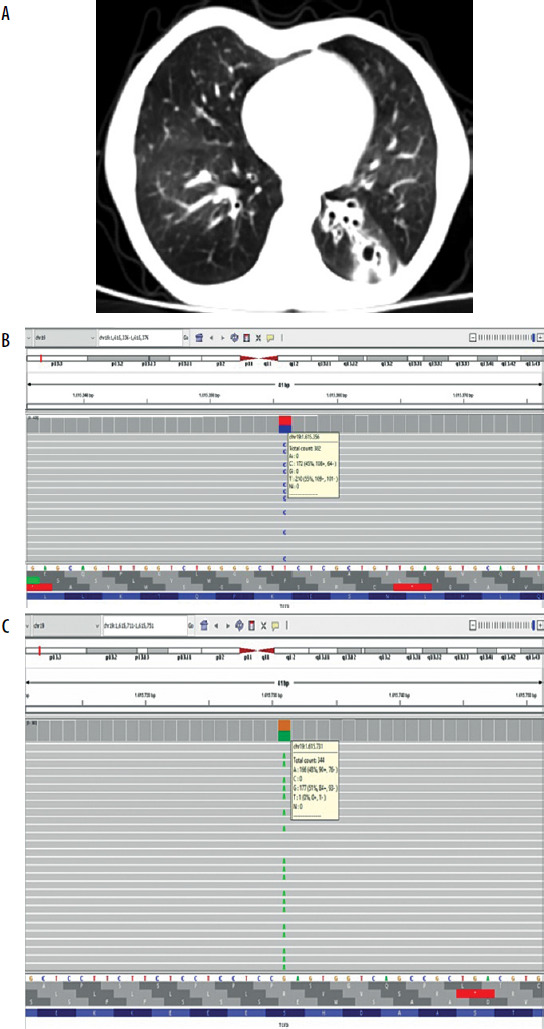
Our second case is a 4-year-old Turkish girl presented with recurrent aphthous stomatitis since the age of one. She had also frequent febrile upper respiratory tract infections (RTIs) at least twice a month and used antibiotics. She has not been admitted to the hospital or intensive care unit. Her older sister is on IVIG due to CVID. The physical examination showed no abnormal findings. Hemogram, serum immunoglobulins, specific antibodies and flow cytometry are shown in Table 1. IVIG therapy was started at 0.4 g/kg once a month with trimethoprim-sulfamethoxazole prophylaxis. The incidence of febrile upper RTIs has decreased and even the mouth sore did not come out after IVIG began. Clinical exome sequencing showed a heterozygous missense mutation, c.1541C>T (p.Ser514Leu), variant of unknown significance, was detected in the TCF3 gene (Figure 1 C). But the same variant was not detected in her older sister. (They did not detect any copy number variations (CNVs) or variant of the other genes associated with CVID in both cases).
DISCUSSION
IEIs causing hypogammaglobulinemia may involve blocking one of the B-lymphocyte maturation steps from pro-B cells to immunoglobulin formation. TCF3 also plays an important role in B-cell development and differentiation [4]. The TCF3 gene has been reported to be associated with IEIs e.g., CVID [5–7]. The TCF3 (E2A) gene encodes two basic helix-loop-helix transcription factors, E12 and E47, through alternative splicing. E12 and E47 are involved in regulation of immunoglobulin gene expression [8, 9]. Homozygous and heterozygous TCF3 gene mutations are a rare cause of agammaglobulinemia.
Autosomal dominant (AD) and autosomal recessive (AR) forms of agammaglobulinemia are described in the literature. AD agammaglobulinemia has been reported in 4 unrelated patients so far. Boisson et al. identified a de novo heterozygous missense mutation, resulting in abnormal protein E555K, specific to the E47 isoform of the TCF3 gene [10]. This mutation resulted in maturational arrest of B cells prior to differentiation into the pro-B cell. These 4 patients had agammaglobulinemia and decreased B cells; the enduring B cells exhibited strong CD19 expression and deficiency of the B-cell receptor [11]. These findings indicated that E47 plays a critical role in implementing the block in the growth of B-cell precursors that lack functional antigen receptors.
In the literature, TCF3 mutations are clinically found to be associated with early-onset, severe, recurrent bacterial infections, bronchiectasis, fibrosis, interstitial pneumonia, neutropenia (severe or intermittent) and growth retardation [10]. A recent case report describes a novel mutation in TCF3 resulting in an AR form of agammaglobulinemia with recurrent pneumonia, chronic diarrhea, and failure to thrive in a 9-year-old female [12]. But another case of a heterozygous mutation in TCF3 presented as selective IgG deficiency and normal B cell counts were reported as well [13]. In another paper, a homozygous nonsense mutation in TCF3 was found in a patient with hypogamma-globulinemia and B-cell acute lymphoblastic leukemia presenting with the absence of all immunoglobulin isotypes and a very low number of peripheral CD19+-B cells [4].
Our patient 1 had recurrent lower RTIs, non-cystic fibrosis bronchiectasis and growth retardation that started at an early age. Patient 2 had frequent febrile upper RTIs instead of lower RTIs and/or bronchiectasis. Nevertheless, they both had hypogammaglobulinemia and B-cell counts were at borderline value or absent (B-cell lymphopenic), according to the reference values [14, 15]. Contrary to the literature, there was no malignancy and/or autoimmune diseases developed in both of our patients [16].
Our patients seemed to have AD agammaglobulinemia and they were shown to have de novo heterozygous missense mutations c.1750A>G (p.Lys584Glu/K584E) and c.1541C>T (p.Ser514Leu/ S514L), variants of unknown significance, specific to the TCF3 gene. The detected variant (K584E) is not seen in the community database (Genome Aggregation Database v2.1.1) and is located in a critically-accepted functional region of the gene [HLH (helix-loop-helix) domain]. When the variant is evaluated with the in silico prediction tool REVEL (rare exome variant ensemble learner), it is predicted that it may have a detrimental effect on protein structure and/or function. Moreover, variant S514L is rare in the community database and is predicted to have a detrimental effect on protein structure and/or function when evaluated with the in silico prediction tool MutationTaster. Briefly, when we evaluate the features of these variants listed here and the clinic tables of our patients together, we thought that these cases should be reported.
As a result, genetic examination should be performed on patients who are followed up with the diagnosis of CVID and have additional findings. Identified genetic mutations have an important place in the clinical follow-up and the phenotype of the disease is determined by case reports. Our case report contributes to the laboratory and clinical presentation of IEI due to TCF3 mutation.










