
INTRODUCTION
Danish geneticist Petrea Jacobsen described in 1973 a three-generation family carrying a terminal deletion at the end of the long arm of chromosome 11 (11q terminal deletion disorder). The 11q deletion is de novo in 85% of reported cases, and in 15% of cases it results from an inherited unbalanced translocation of a familial balanced translocation (carrier parent) or from other chromosome rearrangements [1, 2]. This disorder was later named as Jacobsen syndrome (JS) and it has an estimated prevalence of 1 in 100,000 newborns. Around 200 cases have been reported worldwide so far. The male : female ratio is 1 : 2 [2].
Clinically, JS may include dysmorphic features, congenital heart disease, intellectual disability, cognitive impairment, thrombocytopenia, increased bleeding tendency, and increased susceptibility to infections. The clinical phenotype varies depending on the length, location, and type of chromosomal change [2]. The increased susceptibility to infections is mostly attributed to a problem in B lymphocyte development and humoral immunodeficiency with low memory B lymphocytes. In this case, it causes hypogammaglobulinemia and an impaired response to immunization. Immunoglobulin replacement therapy significantly reduces the burden of infection [3, 4]. In addition, some studies also suggest that low T lymphocyte counts or T lymphocyte dysfunction may be the one of the causes [4, 5]. Another study hypothesized that granular dysfunction of neutrophils may also play a role in this disorder [6].
We present a patient with facial dysmorphism, multiple anomalies, thrombocytopenia, and combined immunodeficiency diagnosed with JS.
CASE REPORT
A 2-year-old Syrian female patient from non-consanguineous family was consulted at the pediatric immunology clinic due to frequent infections and multiple hospitalizations in the pediatric inpatient service. She was born at 37 weeks, 3,000 grams by C/S. She is one of 4 siblings. The patient had been operated on for tetralogy of Fallot when she was 4 months old. Physical examination revealed mental and growth retardation, trigonocephaly, prominent metopic ridge, downslanting palpebral fissures, bilateral Simian line on hands (Table 1), operation scar on the sternum, and a 2–3/6 systolic murmur (Figures 1 A, B). Her weight was 11 kg (3rd p.), and height: 90 cm (15th p.). In the laboratory examinations performed, leukocyte was 5,550/mm3, neutrophil 2,550/mm3, lymphocyte 1,170/mm3, and platelet 54,000/mm3. There was lymphopenia and thrombocytopenia. In the past evaluations, thrombocytopenia and lymphopenia were observed as well. Bleeding time was 5 minutes. Peripheral smear showed normal size and accumulations (lumps) of platelets. Routine evaluations of biochemistry were within normal range. Further immunological evaluations showed IgG: 573 mg/dl, IgA: 32 mg/dl, IgM: 33 mg/dl and IgE: 42.8 IU/ml. Flow cytometry demonstrated lymphocyte subsets of CD3+ cells: 42% (low), CD4+: 17.6% (low), CD8+: 23%, CD19+: 26.7%, and CD16+56+- NK cells: 5.5%. Table 1 compares these values with normal reference ranges, according to the age. Anti-A: 1/8 positive, anti-B titer: negative and anti-HBs titer was 23.71 IU/ml and anti-Rubella: 49.5 S/co (Table 2). Anti- EBV VCA IgG: 15.6 (0–2) AU/ml. Isohemagglutinins and these specific antibody responses to vaccines and recent/previous infections seemed to be low in titer. Latest echography showed an operated modified Blalock Taussig shunt, fully corrected tetralogy of Fallot, right pulmonary artery hypoplasia, and pulmonary valve insufficiency. No pathology related to the liver, kidney, and spleen was observed in the abdominal ultrasound.
Table 1
Comparison of typical clinical features of Jacobsen syndrome with our patient’s characteristics
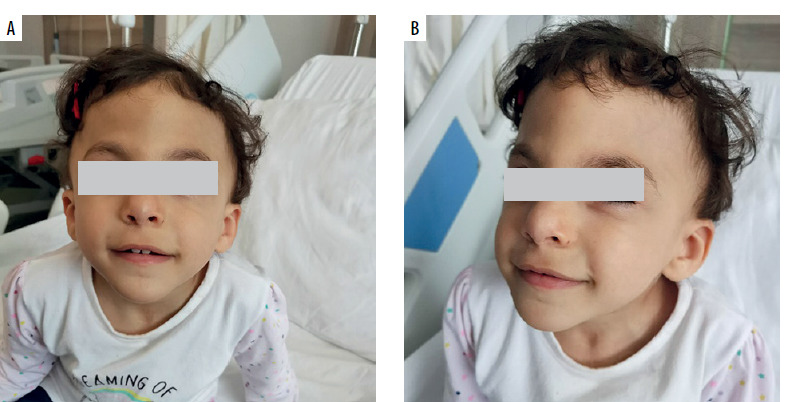
Table 2
Patient’s immunological data at diagnosis
Combined immunodeficiency was diagnosed in the patient who had low IgG, IgA and IgM levels, according to age-specific 95% confidence intervals, together with low CD4+- and CD3+-T cells [7, 8]. Intravenous immunoglobulin (IVIG) replacement therapy was started at 0.5 g/kg once a month. Clinical benefit was seen in the patient with IVIG treatment. Frequent hospitalizations due to infections were decreased.
Briefly, our patient had congenital heart disease (tetralogy of Fallot), thrombocytopenia, combined immunodeficiency, mental retardation, and facial dysmorphism. She has the typical features of JS (trigonocephaly, platelet disorder, heart abnormalities). Brain, skin, gastrointestinal, and urogenital systems are not involved in our patient.
Genetic screening was sent from the patient. As a result of karyotype analysis performed with 72 hours of cell culture from peripheral blood; derivative structure of chromosome 11 was observed as a result of unbalanced transfer of translocation of p25.1 and q24.2 regions of chromosomes 2 and 11; respectively [46,XX,der(11)t(2;11)(p25.1;q24.2)]. Molecular karyotyping was studied from the patient using the “Illumina Infinium HumanCytoSNP-12 v2.1 300K” microarray chip and analyzed with Bluefuse Multi 4.5. In microarray study, deletion finding covering the 11q24.2-11q25 band region in the long (q) arm of the eleventh (11th) chromosome and duplication finding covering the 2p25.3-2p25.1 band region in the short (p) arm of the second (2) chromosome were observed (Figure 2). Although the deletion found in the 11th chromosome partially overlapped with JS, it overlapped with the gene regions thought to be responsible for the immunological and dysmorphic findings of JS (Figure 3). Verbal and written consent was obtained from the patient’s family for sharing the clinic course and pictures.
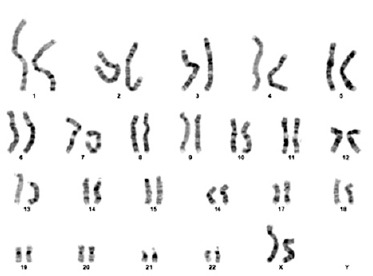
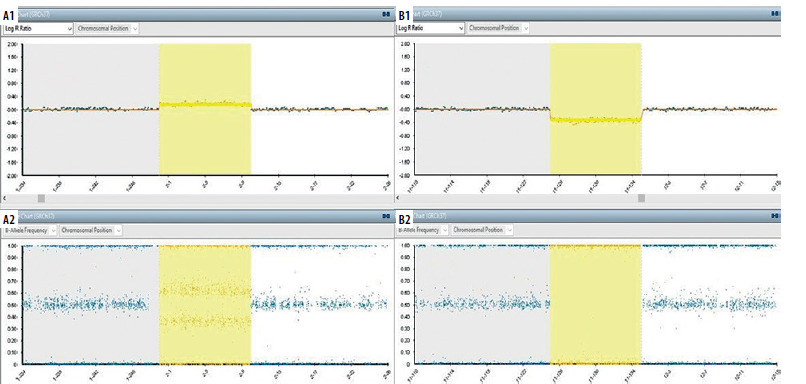
Figure 3
Single nucleotide polymorphism (SNP) array analysis. A1-A2 (arr[Grch37]2p25.3p25.1(72184_10320267). a1: It is the logarithmic expression of the average signal amount for the relevant SNPs (single nucleotide polymorphism). This value, which gives information about the number of copies, indicates the copy gain (duplication) at values above “0”. a2: Shows the ratio of relevant SNP alleles to the total amount of signal. Intermediate values (eg:0.25/0.75) indicate copy gain. B1-B2 (arr[Grch37]11q24.2q25(125026918_134944006)x1). B1: It is the logarithmic expression of the average signal amount for the relevant SNPs (single nucleotide polymorphism). This value, which gives information about the number of copies, indicates the copy loss (deletion) at values below “0”. B2: Shows the ratio of relevant SNP alleles to the total amount of signal. Values of 0 and 1 indicate copy loss
DISCUSSION
Terminal 11q deletion syndrome, called JS (JS; OMIM#147791), is a rare genetic disorder. It affects different systems in the body. With dysmorphic stigmata and intellectual disability; the brain, skin, heart, hematological, gastrointestinal, and urogenital systems are frequently affected [9]. Typical facial features (Table 1) include: skull deformities (macrocrania, prominent forehead, facial asymmetry, trigonocephaly), ocular hypertelorism, downward sloping palpebral fissures, strabismus, palpebral ptosis, sparse eyebrows, epicanthal folds, straight or prominent nasal bridge, short nose, anteverted nostrils, broad nasal bridge, small ears, low ears turned backward, malformed outer ears, long philtrum, straight philtrum, V-shaped mouth, thin upper lip, and retrognathia [9]. Trigonocephaly is a skull shape abnormality in which the forehead appears triangular when viewed from above. It usually occurs with premature closure of metopic sutures. Trigonocephaly causes a characteristic facial appearance and increases the probability of diagnosis. In our patient, trigonocephaly is remarkable (Figures 1 A, B) and the comparison of our patient with the features of the JS reported in the literature is given in Table 1.
Congenital heart malformations, most of which require medical treatment and/or surgical repair, occur in 56% of cases with JS. Cardiac malformations have been reported in 95% of the children who died [10]. They are most common in 2/3 of patients with congenital heart defects; ventricular septal defects or left heart obstructive malformations (aortic or mitral valve abnormalities, aortic coarctation, Shone’s complex or hypoplastic left heart syndrome) [11]. Our patient had tetralogy of Fallot.
The most common laboratory finding is thrombocytopenia [2]. Many JS patients are born with thrombocytopenia and platelet dysfunction, called Paris-Trousseau syndrome (PTS) [11]. This thrombocyte abnormality has a high penetrance in JS. It affects at least 88.5% of people with a hemizygous loss of the FLI-1 gene [2]. Heterozygous loss of the FLI-1 gene is also associated with Paris-Trousseau thrombocytopenia in JS [1]. It is known that the diagnosis of PTS has important clinical implications. These children are at potentially life-long risk for life-threatening bleeding complications, especially in surgeries involving significant bleeding. Some researchers assume that anyone whose 11q deletion includes the FLI-1 gene is diagnosed with PTS [1]. Deletion of at least 3 of the 4 platelet function critical genes (FLI-1, ETS-1, NFRKB and JAM3) is shown to be necessary for thrombocytopenia. Our patient has loss of the 3 of 4 genes (ETS-1, FLI-1 and NFRKB) and thrombocytopenia. Therefore, she is followed closely in terms of bleeding complications.
Jacobsen syndrome was detected by us while investigating the genetic cause in our patient who had immunodeficiency, dysmorphic findings, and multiple anomalies. No pathological or probable pathological variants were detected in the immunodeficiency panel performed by next-generation sequencing analysis. The derivative structure of chromosome 11 was observed in karyotype analysis from peripheral blood. Thereupon, karyotype analysis was performed with the microarray method. A deletion finding of approximately 9.9 Mb covering the 24.2-25 band region was observed in the long (q) arm of the eleventh (11th) chromosome of our patient. This region contains 76 genes including TIRAP (Toll/interleukin-1 receptor domain containing adapter protein), ETS1 (erythroblast transformation-specific 1), FLI-1 (friend leukemia virus integration-1), NFRKB (nuclear factor related to kappa B-binding protein), THYN1 (thymocyte nuclear protein 1), SNX19 (sorting nexin 19), ARHGAP32 (Rho GTPase activating protein 32), and JAM3 (junctional adhesion molecule 3), which are known to be immunologically active [4, 12]. In the deletion of our patient, both the BARX2 (BarH-like homeo box gene 2) gene responsible for dysmorphia and the genes with immunological function were lost [12]. Similarly, Blazina et al. reported a 16-year-old JS patient with combined immunodeficiency in whom these immunologically active genes were also deleted [5].
Genetically, focus is on the transcription factors ETS1 and FLI1, both located on 11q24.3. Mice with deletions in the ETS1 gene appeared to have significant defects in T, B, and NK cell development and spontaneous apoptosis of T lymphocytes is increased, providing a prediction that this would produce an immunological phenotype [13]. However, studies in humans and genetically engineered mice suggest that ETS1 (specific for E26 transformation 1) is the cause of congenital heart defects in JS, but the underlying molecular and cellular mechanisms are unknown [14]. FLI1 is a member of the ETS gene family that shares a DNA-binding domain called ETS domain, which is responsible for sequence-specific DNA recognition of target promoters. This domain has a role in development of B and T lymphocytes [6]. Our patient had congenital heart disease and T-cell subset deficiency since the ETS1 gene was deleted.
The first immune defect reported in JS patients was antibody deficiency [15, 16]. Comprehensive immunological evaluation has not been done much as these patients did not have severe infections. In a large prospective study involving 110 patients with terminal 11q deletion, no life-threatening or opportunistic pathogenic infections were reported. Upper respiratory tract infections were common in these patients, with recurrent otitis media and/or sinusitis episodes being the most common [2]. The absence of significant T cell defects and immunological evaluation in most of the JS patients reported in the literature led to the thought that JS predominantly causes antibody deficiency. However, there are cases whose immunodeficiency was evaluated in detail and combined immunodeficiency was also found [5, 17]. In a case report published by Blazina et al., the patient suffered from recurrent purulent otitis media and mostly upper respiratory tract infections since the kindergarten period. Immunological examination at that time was consistent with antibody deficiency. However, recurrent viral and bacterial upper respiratory tract infections and lobar pneumonia of the 12-year-old patient continued. A comprehensive reassessment of the 16-year-old patient showed decreased B-cells and reduced T-cell subtypes. The patient was diagnosed with combined immunodeficiency [5]. In addition, a 12-year-old pediatric JS patient and low IgG, IgM, T helper, T cytotoxic, and B- cells was reported by Fernández-San José et al. [17]. Another case is the definition of late-onset combined immunodeficiency in a 45-year-old female patient diagnosed with JS [4]. Our patient had also hypogammaglobulinemia and T lymphocyte subsets. Our patient predominantly suffered from recurrent lower and upper respiratory tract infections. But there were also several pediatric intensive care admissions in her past medical history. She is still being followed up with the diagnosis of JS with combined immunodeficiency.
Prenatal diagnosis of 11q deletion is possible by amniocentesis or chorionic villus sampling and cytogenetic analysis. Management is multi-disciplinary and requires evaluation by a general pediatrician, pediatric cardiologist, neurologist, and ophthalmologist [9, 18]. Cardiac malformations can be very severe and require heart surgery in the neonatal period. Newborns with JS may have difficulties in feeding and tube feeding may be necessary. Special attention should be devoted due to hematological problems. About 20% of children die during the first two years of life, most commonly related to complications from congenital heart disease, and less commonly from bleeding. For patients who survive the neonatal period and infancy, the life expectancy remains unknown [9]. Our patient had been operated on for tetralogy of Fallot when she was 4 months old.
Differential diagnoses include Kabuki, Turner and Noonan syndromes, and acquired thrombocytopenia due to sepsis [9].
CONCLUSIONS
Jacobsen syndrome is a rare genetic cause of syndromic primary immunodeficiency. Patients with multiple anomalies and immunodeficiency should be investigated for genetic mutation. Early detection of immunodeficiency can reduce the frequency and severity of infections. Patients with JS should be examined in terms of immunodeficiency as well as additional findings. Large cohort studies are needed to more precisely define the pathophysiology of immunodeficiency in JS patients.










