
INTRODUCTION
Antipsychotics were discovered in the late 1950s as part of the process of improving anesthetic agents. They proved ineffective as anesthetic drugs but, surprisingly, attenuated psychotic symptoms in patients. In the 1960s and 1970s the primary mechanism of antipsychotic action was determined to be the antagonism of dopamine receptors (DRD2) and these drugs were referred as “typical”. In late 1980s and 1990s “atypical” or “second- generation” antipsychotics were introduced to improve the treatment of negative and cognitive symptoms of schizophrenia and to alleviate extrapyramidal side effects (clozapine). Atypical antipsychotics turned out to be multi-action agents, blocking not only DRD2. They induce less severe extrapyramidal side effects but, on the other hand, were associated with weight gain and obesity in patients [1]. They all have ability to modulate intra-cellular pathways, metabolism, the structure or even apoptosis of cells, which probably corresponds to the impairments associated with psychiatric disorders but also with their therapeutic potential and side effects. Despite the use of antipsychotics to treat psychotic and affective disorders over the last several decades, knowledge about their molecular actions is still incomplete. The aim of this review is to present the current and basic knowledge about the mechanisms of antipsychotics’ functioning at the intracellular level, in light of their clinical efficacy and side-effects. The direct and indirect molecular mechanisms of antipsychotics will be discussed on the basis of three stages of signaling: intracellular pathways, and epigenetic (DNA methylation and histone modification) and post-transcription (microRNA) processes.
INTRACELLULAR SIGNALING PATHWAYS
Intracellular signaling is necessary for the regulation of gene transcription. The common intracellular mediators include cyclic adenosine monophosphate (cAMP), AKT pathway known also as protein kinase B (PKB)/glycogen synthase kinase-3 (GSK-3) and the mitogen-activated protein kinase (MAPK) pathway. All antipsychotics act on the DRD2 receptors, which are Gi/o type G-protein-coupled receptors; their main downstream signaling pathway is adenylate cyclase-cAMP protein kinase A (PKA). The influence of typical and atypical antipsychotics on cAMP and PKA is complex and region-dependent, probably due to their affinities for different G-protein-coupled receptors stimulating adenyl cyclase signaling [2]. The majority of antipsychotics blocking DRD2 receptors increase cAMP production by removing the tonic inhibition of dopamine transmission by Gi/o proteins on adenylate cyclase activity. The increase in cAMP production activates PKA and increases PKA-dependent phosphorylation of DARPP-32 (dopamine- and cAMP- regulated phosphoprotein of molecular weight 32 000). DARPP-32 is a neuronal phosphoprotein that displays a regional distribution in the mammalian brain very similar to that of dopamine-containing nerve terminals, being highly concentrated in the basal ganglia. The main role of DARPP-32 is the integration of responses to natural reinforcing stimuli, with a focus on food as a natural reinforcer [3]. In the prefrontal cortex of patients with schizophrenia, levels of DARPP-32 have been shown to be significantly decreased [4]. Typical and atypical neuroleptics differ in their influences on the PKA pathway. Treatment with haloperidol has been shown to increase PKA activity by enhancing its mRNA and protein synthesis in the rat striatum, while clozapine reduced these parameters in the striatum, hippocampus and frontal cortex of rats [5]. The administration of halo peridol and clozapine induces DARPP-32 phosphorylation at the TH34 site, converting it into an inhibitor of proteophosphates 1 (PP1) in mouse dorsal stratum, which probably affects gene transcription and synthesis of protein like cAMP (cAMP response element-binding protein), which is a cellular transcription factor binding to certain DNA sequences called cAMP response elements (CRE) [6].
The next example of intercellular signaling is the AKT/ glycogen synthase kinase 3 (GSK-3) pathway (Figure I). GSK-3 is a serine-threonine protein kinase and is negatively regulated by the phosphorylation of its serine residues by AKT (known as protein kinase B). The role of kinase enzyme is to transfer a phosphate group from a high-energy donor molecule to residues (serine, threonine or tyrosine) on the recipient molecule. The inactivation of GSK-3 via phosphorylation by AKT has important consequences for the functioning of neurons. GSK-3 is implicated in glucose metabolism, neurodevelopment, differentiation and development, intracellular trafficking, apoptosis, regulation of gene transcription, and synaptic plasticity in the brain [7, 8]. SNPs (signal nucleotides polymorphisms) of GSK-3β are described to be risk factors for schizophrenia. The synthesis of glycogen is regulated by two subunits of GSK-3 (α and β) [9]. AKT plays a role in cognition through its modulation of long-term potentiation, synaptic plasticity and, in consequence, working memory [10]. Three isoforms of AKT have been detected. AKT1 is the isoform most associated with schizophrenia; its reduced levels have been revealed in the brains (hippocampus and frontal cortex) of patients suffering from schizophrenia, whereas increased signaling of AKT1 was observed in rodent neurons in response to acute antipsychotic treatment. Genetic have studies revealed that AKT1-mediated regulation of neuronal cell size and volume, and its polymorphism was associated with brain volume reduction in the caudate bilaterally and right prefrontal cortex [11]. AKT2 is implicated in the regulation of metabolism through insulin-regulated glucose homeostasis and AKT3 is involved in postnatal brain development [12].
Figure I
Intracellular signaling cascades modulated via dopamine receptor D2 (D2R) transmission. Binding of dopa-mine to the D2R receptor leads to recruitment of β-arrestin 2 along with AKt and phosphatase PP2. PP2 dephosphorylates and inactivates AKt what in consequence increased GSK-3 activity. GSK-3 creates complex with β-catenin, APc and Axin. the alternative pathway with mtORc1 is a protein complex that function as a nutrient/energy/redox sensor and controls protein synthesis through the direct phosphorylation of two key targets, S6K1 and S6rp
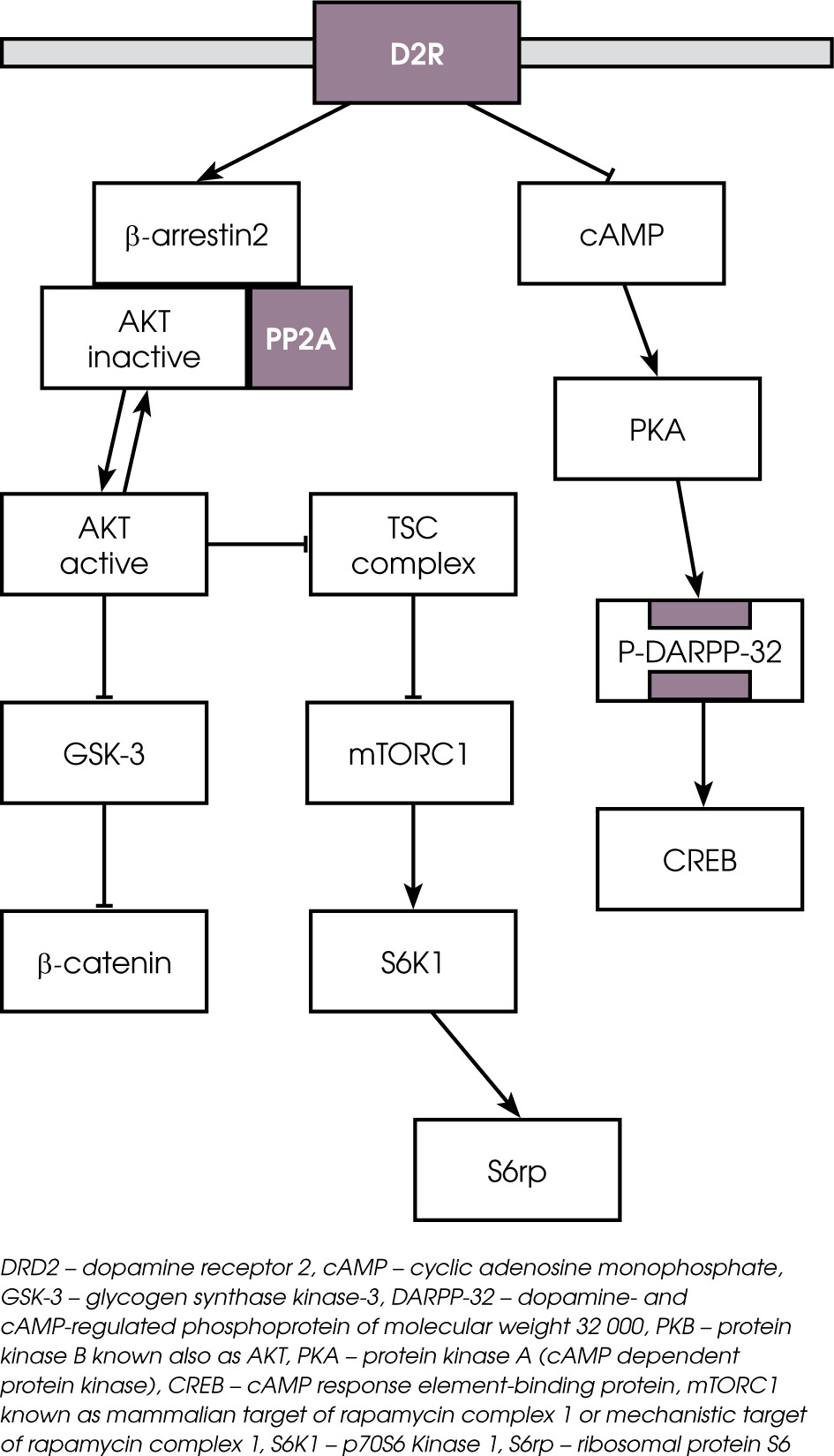
Activation of the postsynaptic D2 receptor by dopa-mine (DA) leads to formation of the complex of molecules consisted of by binding β-arrestin 2 (scaffolding protein), AKT and phosphatase 2A (PP2A). The role of this complex is to desensitize the D2 receptor, which subsequently leads to its internalization. Thus, the activation of dopamine D2 receptors by dopamine leads to the dephosphorylation of the phosphorylated and inactivated AKT complex by phosphatase PP2 (an enzyme that removes a phosphate group from its substrate). Inactivated AKT complex indirectly leads to increased GSK-3 activity, as active AKT exerts inhibiting influences on both isoforms of GSK-3 (α and b) [7]. The G-protein-independent intracellular cascade β-arrestin 2-PP2A-AKT plays an important regulatory role in the modulation of neurons by DA or antipsychotics.
Antipsychotics acting as DRD2 antagonists inhibit the assembly and activation of the complex of β-arrestin, PP2A and AKT. Next, the prevention of β-arrestin 2/PP2A recruitment leads to phosphorylation and the activation of AKT and, in consequence, the AKT signaling is not inhibited [9, 13]. AKT is a convergent antipsychotic signaling mechanism. Although AKT pathways are modulated by multiple first and second generation antipsychotics, the extent of impact varies between different antipsychotic drugs. Masari et al. demonstrated that antipsychotics such as haloperidol, risperidone, ziporasidone, clozapine, olanzapine and aripiprazole all prevented the binding of β-arrestin 2 to DRD2 induced by the administration of a DRD2 agonist in cell culture [14]. Antipsychotic drugs like haloperidol, risperidone, olanzapine, cloza-pine, quetiapine, or ziprasidone have been demonstrated to increase the phosphorylation of GSK-3 by AKT, which inhibits GSK-3 activity in different brain regions [15, 16]. An increased level of AKT in the prefrontal cortex, hippocampus and striatum was described in of rats treated with olanzapine [17]. Other studies also showed that both classes of antipsychotics have similar effects on the AKT-GSK pathway. Some neuroleptics (clozapine, olanzapine) produce the phosphorylation of AKT and GSK, whereas others (haloperidol) induce the phosphorylation of only AKT, which leads to further phosphorylation of GSK-3. Interestingly, the newer antipsychotics, such as aripiprazole, activate AKT-GSK-3β signaling [18], which is similar to the activation of AKT-GSK-3β signaling by mood stabilizers such as lithium [9]. It is also worth noting that lithium has been shown to disrupt the β-arresting 2/ Akt/PP2A complex, preventing the dopamine-induced dephosphorylation of AKT, which was expressed clinically as the blocking of amphetamine-induced locomotion (via dopaminergic transmission) [19]. The activation of AKT is upstream of multiple signaling cascades involved in transcription, protein synthesis, metabolism and other cellular processes. For example, the AKT-mTORC1 (mammalian target of rapamycin complex 1, or mechanistic target of rapamycin complex 1) pathway has been proven to be another site of antipsychotic modulation. This is a protein complex that functions as a nutrient/energy/redox sensor and controls protein synthesis. Bowling demonstrated that acute antipsychotic treatment induces the action of AKT-mTORC1 protein synthesis pathway, resulting in immediate changes in protein synthesis [20].
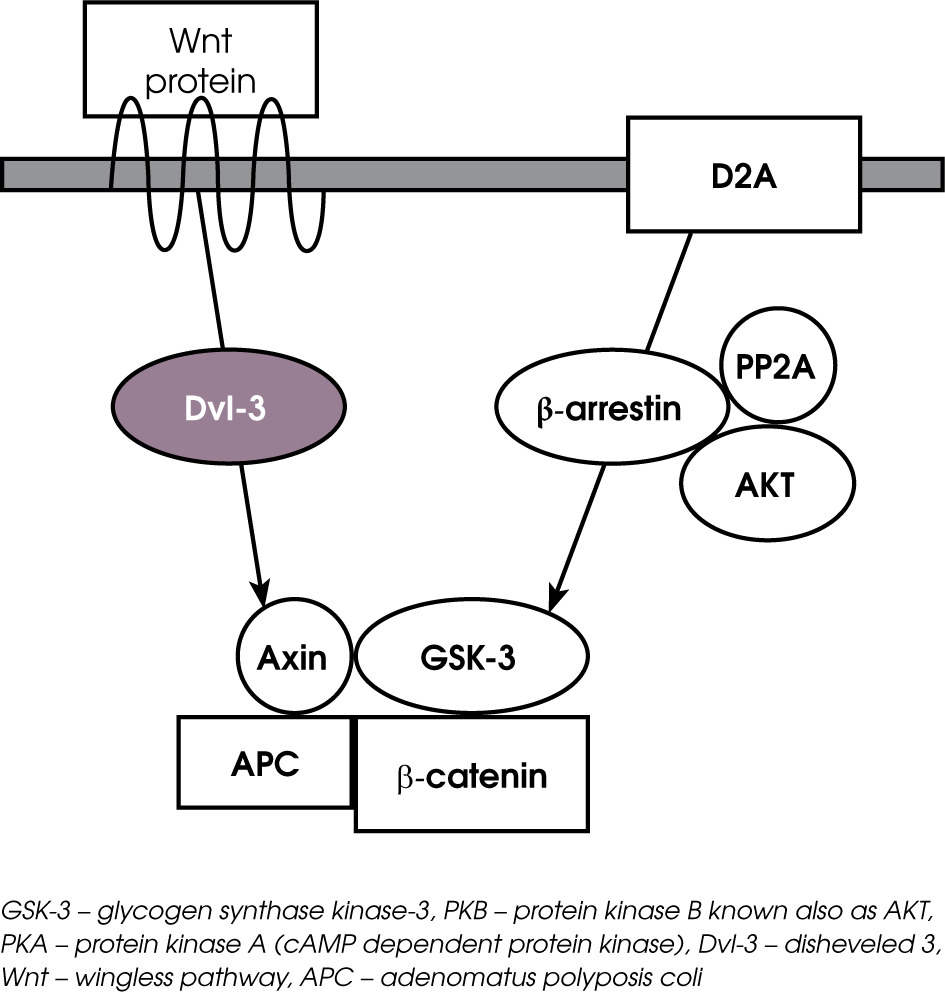
The mechanism of intracellular signaling associated with AKT/GSK-3 is the Wingless (Wnt) receptor pathway (Figure II). These two pathways of intercellular signaling work in parallel and intersect with each other. Wnt signaling is a group of signal transduction pathways which begin with proteins that pass signals into a cell through surface receptors and next to the Dishevelled protein (Dvl) inside the cell. Members of the Wnt protein family are secreted glycoproteins. The Wnt pathways are comprised of three different complexes: the “canonical” pathway involving GSK-3 and β-catenin regulating gene transcription, the “noncanonical” cell polarity pathway (PCP) that regulates the cytoskeleton responsible for cell shape, and the “noncanonical” Wnt/calcium pathway that regulates calcium transport inside the cell. Thus the Wet signaling network is associated with cell differentiation, proliferation and migration, playing a significant role in processes of embryogenesis and carcinogenesis. Abnormalities in Wnt pathways have been described in type II diabetes, tissue regeneration in adult bone marrow, and abnormal brain development [21]. Wnt signaling has also been described as neurodevelopment abnormality in schizophrenia patients [22]. Dishevelled-3 (Dvl-3) is the protein associated with the D2 dopamine receptors and with Wnt receptors. Antipsychotics may act on Dvl-3 via D2 dopamine receptors to initiate a cascade of downstream changes involving Axin, GSK-3 and β-catenin [23]. Axin and GSK-3 form a complex along with APC protein (adenomatous polyposis coli, a tumor suppressor), which is responsible for the phosphorylation and destruction of β-catenin. β-catenin is a protein accumulating in the nucleus and regulating gene-transcription through interactions with DNA transcription factors. In the absence of Wnt receptor activation, β-catenin is continuously phosphorylated by GSK-3, leading to degradation. The binding of ligands to Wnt receptors leads to the phosphorylation of Wnt-receptor related proteins (LRP5 or LPR6) and activation of the disheveled intra-cellular protein (Dvl), enabling the separation of GSK-3 from β-catenin. Next, the phosphorylation of GSK-3 by Akt prevents β-catenin phosphorylation by GSK-3 and allows β-catenin to migrate to the nucleus and regulate gene transcription. A significant increase in Dishevelled 3 (Dvl-3), β-catenin and GSK-3β been revealed after chronic treatment with clozapine, haloperidol, risperi done in rat ventral midbrain, medial prefrontal cortex and striatum [24]. Chronic treatment with clozapine and haloperidol has been shown to increase Wnt-5a, then the levels of Dvl-3, Axin and AKT phosphorylating GSK-3β in the rat frontal cortex [23]. Both clozapine and haloperidol induce the phosphorylation of AKT and GSK-3β [16]. In the group of antipsychotics, clozapine shows an outstanding profile of influence on the Wnt and AKT pathway as it induces phosphorylation of GSK-3β and AKT but being the only drug that increases both phosphorylation and the amount of Dvl in the rat frontal cortex [16]. Moreover, clozapine has been shown to enhance GSK-3β signaling via the activation of the Wnt pathway but not via AKT signaling [25]. The above facts suggest that antipsychotics the first and second generations differently regulate the AKT and Wnt pathways, which is probably dependent on their relative D2 or 5-HT2 receptor antagonism. The unique effects of clozapine might be related to its unique profile of action.
Figure II
The Wnt intracellular signaling cascade. When Wnt receptor is not activated, β-catenin is phosphorylated by GSK-3 and degraded. In consequence, β-catenin gene transcription is inhibited. Binding a ligand to Wnt receptor results in the formation of complex with Dvl, APc, Axin GSK-3 and AKt. Inactivation of GSK-3 by AKt 1 prevents β-catenin phosphorylation and its degradation. In consequence, β-catetin accumulates in the nucleus and regulates transcription
One of the clinical significances of Wnt/β-catenin signaling is regulation of osteoblast apoptosis, which might contribute to antipsychotic-induced osteal side effects, like decreased bone mineral density and fractures [26]. Wnt/β-catenin signaling is an important bone homeostasis modulator through the promotion of mesenchymal stem differentiation into osteoblasts. This study proved that antipsychotics induced apoptosis of osteoblasts through Wnt/β-catenin signaling suppression, which has been suggested to be a novel mechanism apart from the elevation of prolactin secretion caused by such drugs.
In conclusion, disruption of AKT, GSK-3 or Wnt signaling in the brain has been demonstrated to be involved in brain disorders like schizophrenia, and its restoration through the use of antipsychotics may play a significant role in clinical improvement. For example, reduced AKT level in some regions of the brain in schizophrenia patients leads to enhanced of GSK-3 activity, and in consequence the degradation of the gene transcription modulator – β-catenin. In addition, the increased expression of the Wnt-1 a molecule in the Wnt pathway has been described in the hippocampus of subjects with schizophrenia [27], and an association between genes in the Wnt signaling pathway and psychotic subtypes of bipolar disorder has been demonstrated [28].
The last intracellular mechanism described here is the MAPK (mitogen-activated protein kinase) pathway, which is important in a number of neuronal functions such as regulation of gene expression, protein synthesis and receptor modulation. The above processes contribute to synaptic plasticity, learning and memory. Antipsychotic drugs may modulate the MAPK pathway in different ways. Typical and atypical antipsychotics exert different impacts on extracellular signal-regulated kinases 1/2 (ERK 1/2) phosphorylation in the brain. For example, a single injection of haloperidol increases the phosphorylation of ERK 1/2 and Elk1 (Elk-1, a member of the Ets domain transcription factor family) in the dorsal striatum in mice, whereas clozapine administration impairs these phosphorylations [6]. Moreover, the research demonstrated that clozapine but not olanzapine or halo peridol induced initial inhibition and subsequent activation of cortical and striatal ERK 1/2 and p90RSK (p90 ribosomal S6 kinase) phosphorylation in mice. This clozapine- induced ERK 1/2 activation is mediated by the epidermal growth factor receptors (EGF), which demonstrates the unique action of clozapine in comparison to other anti-psychotics [29, 30].
EPIGENETICS
Epigenetics is a new area of study exploring the influences of environment on phenotypic outcome during ontogenetic development. Epigenetics involves functional changes to genetic expression without any changes in gene sequences. Many recent studies have shown that epigenetics are involved in the etiology of many somatic and mental disorders [31]. Environmental influences on gene expression occur due to the modification of histone, changes in the methylation level that regulates transcription, translation and protein synthesis and RNA based mechanisms such as microRNA. The analysis conducted by Burghardt demonstrated that the pharmacoepigenetic literature on antipsychotic use gives inconsistent and variable results and there is an urgent need to conduct prospective surveys [32]. The epigenetic characterization of an organism is expressed by of the genomic methylation at the broad level as global DNA methylation, reflecting the methylation status of total genomic content within a given sample. The next level of epigenetic changes is the gene-specific methylation critical regulatory regions like CpG islands and promoter areas. In line with the result of the review by Burghardt, antipsychotic drugs have been shown to influence specific epigenetic mechanisms. The epigenome-wide association studies showed that anti psychotics induce methylation changes within a wide number of genes and pathways like the WNT/β-catenin pathway, in lipid metabolism and in immune-related, neurogenesis, cell adhesion, respiration and mitochondrial activity [33]. The gene-specific studies concern seve ral specific genes, but among them only the genes for COMT (catechol-O-methyltransferase), 5-HTR2A (5-hydro xytryptamine [serotonin] receptor 2A) and 5-HTT (5-hydroxytryptamine/serotonin-transporter) have been described in more than one study. Other studies have shown that there were no uniform or replicable changes in methylation (increase or decrease) in anti-psychotic-treated patients, mainly due to the variability of the sample population, poorly defined antipsychotic exposure, and the use of different methodologies of analysis. Studies also revealed that peripheral blood DNA methy lation is less correlated with brain DNA methylation, whereas saliva appears to have a higher correlation with the brain methylation level [34].
Histones are the primary protein components of euka ryotic chromatin, which play a role in gene regu lation [35]. Histone modifications were detected in samples from patients treated with antipsychotics. Studies reported that long-term clozapine and risperidone but not haloperidol treatment strongly decreased H3 acety lation at the promoter site of metabolic glutamate receptor 2 (mGlu 2) in mouse frontal cortex [36]. This corresponds with the findings of decreased histone H3 acetylation at the mGlu2 promoter and increased expression of HDAC2 (histone deacetylase) in the frontal cortex of schizophrenia patients treated with atypical antipsychotics [36]. Another mechanism of antipsychotic action associated with histone modification is Hdac2 (histone deacetylase) transcription regulation. Endogenous Hdac2 activity plays a critical role in cognitive performance as an enhanced level of Hdac2 leads to restraint of structural and functional synaptic plasticity [37]. The family of transcription factors involved in the regulation in synaptic plasticity, inflammation and immune responses is NF-κB. Increased activity of NF-κB leads to its binding to the Hdac2 promoter and enhancement of Hdac2 transcription. It has been found that known that chronic antipsychotic medication leads to a decline of cognitive abilities in rodent models and cognitive impairment in schizophrenia patients, though they do lead to a significant reduction of psychotic symptoms. Chronic atypical antipsychotic drug treatment led to elevations in Hdac2 promoter activity within the frontal cortex through a linear signaling pathway that involves the 5-HT2A- receptor. A study by Ibi et al. revealed similar effects after chronic treatment with the atypical antipsychotics like clozapine, risperidone, quetiapine, and sulpiride but not with the typical antipsychotic haloperidol. They also demonstrated that atypical antipsychotic clozapine but not haloperidol induced detriment in synaptic remodeling and cognitive processes, which was associated with 5-HT2A receptor-dependent augmentation of Hdac2 epi-genetic function [38]. It would be necessary in further research to find out whether pharmacological inhibitors of this pathway (5-HT2A) improves brain plasticity and cognition in patients treated with atypical antipsychotics. Clini cal studies demonstrate that drugs such as valproate, one of whose functions is to act as a nonspecific histone deacetylase (HDAC) inhibitor, are efficacious when given chronically in combination with atypical antipsychotic drugs, which suggests HDAC2 as a promising new target for schizophrenia treatment [39].
Studies of global DNA methylation have revealed a positive correlation of methylation with clozapine treatment, and a negative correlation with diagnosis. Higher methylation levels were demonstrated in antipsychotic- treated patients [40] and a higher level of methylation was detected in patients treated with haloperidol compared to other antipsychotics, at the similar level as in healthy control group [41]. On the other hand, other studies did not prove any associations between antipsychotics and methylation [42]. Some research has demonstrated that haloperidol treatment is associated with higher levels of global DNA methylation in the leucocytes of patients with schizophrenia [41], and with increase in the phosphorylation and acetylation of histone H3 in the mouse striatum [43]. It has also been shown that haloperidol differently affects DNA methylation in various rat tissues depending on sex. Studies conducted by Shimabukuro et al. showed statistically significant differences in the methylation content of leukocyte DNA in rats (males more than females), which was also consistent with previous results demonstrated in humans [44]. The apparent paradox of a lower methylation state in females may be partly explained by the global hypomethylation of the inactive X chromosome. However, there was no difference in the mC (genomic 5-methylcytosine) content between males and females in either the brain or liver DNA in drug-free control groups. After haloperidol treatment, increased global DNA methylation in both males and females was discovered in the liver, but the difference was statistically significant in males only. In the brain, haloperidol treatment resulted in a decrease of methylation in females, and the decrease was statistically significant. Thus, the differential effects of haloperidol on DNA methylation in various tissues and the sexes suggest that specific hormones may play a role in haloperidol’s modification of the methylation state of DNA, and that processes are complex [45].
Epigenome-wide methylation studies have shown antipsychotic-induced methylation, associated with a wide range of cellular processes. Insulin resistance induced by antipsychotic treatment is associated with gene-specific DNA methylation, in particular the Wnt/β-catenin pathway [46]. The studies of Kinoshita et al. revealed DNA methylation decrease in genome sites responsible for the expression of cell adhesion molecules in patients after a year of clozapine treatment [47]. The studies also indicate that antipsychotics may directly modify gene-specific methylation. Clozapine and sulpiride, but not haloperidol and olanzapine, induced the demethylation of the methylated genes of reelin and GAD67 (glutamic acid decarboxylase 67), as a low expression of these genes has been found in the brains of subjects with schizophrenia and bipolar disorder [48, 49]. Studies conducted by Melka et al. [50] revealed epigenetic changes in some brain regions induced by olanzapine. Significant differences in methylation in genes related to dopamine expression were described in the hippocampus, cerebellum, and liver, for example: significantly increased methylation at the dopamine-DARPP-32 pathway and decreased methylation at CDc42 gene in hippocampus (cell division control protein 42 homolog – a protein involved in regulation of the cell cycle). This might be particularly relevant to the clinical potential of olanzapine as Cdc42 is a signaling protein important for the reorganization of actin cytoskeleton and morphogenesis of cells, playing a role in synaptic plasticity, learning and memory processes. In the cerebellum, increased methylation was seen in ephrin- receptor signaling, the role of which is to mediate a variety of cell-cell interactions during embryonic development and during adulthood in the process of long term potentiation, angiogenesis and oncogenesis [50]. Studies by Abdolmaleky showed that lifetime antipsychotic use was correlated with decrease in methylation of DTNBP1 (dystrobrevin binding protein 1) [51] and the 5-HTT (serotonin transporter) gene-site [52], but increased methylation of the HTR2A site in comparison to drug-free patients [53]. Burghardt [54] demonstrated increased DNA methylation observed for AKT1, AKT2 but not in the AKT3 gene site in patients treated with antipsychotics (31% quetiapine, 25% risperidone, 19% olanzapine, 13% asenapine, and 13% aripiprazole) versus patients treated with mood stabilizers (43% lamotrigine, 43% lithium, and 14% valproic acid) and this trend was observed between AKT methylation and insulin resistance.
In conclusion, it might be suggested that gene methylation changes in tissues (mainly in skeletal muscle) and the expression of key insulin-signaling protein (AKT) may be responsible for the development of insulin resistance during antipsychotic treatment. Results from other studies on the role of antipsychotics in methylation status are inconsistent, as D’Addario revealed no effects of antipsychotic use in patients with schizophrenia [55]. An analysis taking into account correction for multiple testing also revealed no associations for any medication [56]. Contrarily, Venugopal et al. described increased methylation to the level similar to the control group in patients treated with antipsychotics [57].
PROTEOMIC AND TRANSCRIPTOMIC STUDIES
The hypothesis for the origin of schizophrenia mostly concerns developmental and functional abnormalities in brain regional connectivity, neurotransmitter systems of neurons, and the activity of oligodendrocytes and the myelinization process. Antipsychotics have been shown to influence many intracellular processes in neurons and oligodendrocytes, by modulating synthesis and processes like translation initiation, translation elongation and folding in the class of proteins associated with metabolism, protein synthesis, the cytoskeleton, cell adhesion and synaptic activity. Reduced levels of transcripts in blood and olfactory cells has been revealed in patients with schizophrenia in comparison to control subjects [58]. The upregulation of proteins involved in the synthesis of ribosomal proteins and tRNAs is one of the cellular process targeted by antipsychotics. Studies conducted by Kumarasinghe [59] demonstrated that antipsychotics normalized aberrant AKT1 but not DISC1 (disrupted- in-schizophrenia 1) protein transcript levels in mono-nuclear cells of schizophrenia patients compared with healthy volunteers. The same study also revealed that anti-psychotics restored the physiological levels of ribosomal proteins and tRNA (transfer RNA) synthesizing protein in a group of patients. Bowling et al. described, in their studies, the role of antipsychotics in protein synthesis due to modification of mTORC 1 signaling and changes in ribosomal proteins within the first hours of treatment [19].
The group of proteins involved in respiratory and metabolism processes is also a target for antipsychotics. Mitochondria-related oxidative stress and damage were evident in the prefrontal cortex tissue of patients suffering from schizophrenia, including decreased mitochondrial size and altered cellular distribution [60]. Antipsychotic treatment has been shown to alter the transcription and protein abundance of several specific genes indicated in mitochondrial and metabolic defects. Numerous changes in the level of metabolism-related transcripts and proteins such as DISC1 (disrupted-in-schizophrenia), optic atrophy 1 (OPA 1), glutamate dehydrogenase 1 (GLUD1) and AKT1 have been revealed after acute and chronic antipsychotic treatment [61-63]. Moreover, the altered expression of proteins involved in glycolysis and glucose metabolism was described in cortical synaptoneurosomes of rats treated with antipsychotics for 4 weeks, which suggests alterations in mitochondrial and energy metabolism [64].
The genes involved in cytoskeleton processes like micro-tubule dynamics, dendritic branching, and spine dynamics are also the area affected in schizophrenia and targeted by antipsychotics. One of these cytoskeletal proteins is micro tubule-associated protein 2 (MAP2). The expression of the MAP2 gene, usually decreased in human patients with schizophrenia, is increased at the transcript and proteome level during treatment with antipsychotics [65]. Rodent have studies revealed an increase in MAP2 gene transcripts in the brain following antipsychotic treatment [61]. Cell adhesion processes regulated by genes, coding cell adhesion molecules and synaptic stability are also the shared mechanisms underlying disease and treatment [66, 67]. Analysis of prolonged antipsychotic treatment and protein synthesis (ribosomal, chaperone and tRNA-related proteins) demonstrated that cytoskeletal protein expression may be induced within a day of treatment and maintained. In addition, other cytoskeletal proteins and rodent neural progenitor cells (NPCs) such as dynamic 1 (DNM1), glial fibrillary and acidic protein (Gfap) have been demonstrated to be increased during antipsychotic treatment [68]. Finally, alterations in the abundance of cell-adhesion and synaptic-activity proteins have been demonstrated as being due to antipsychotic treatment. The altered proteins include: NCAM1 (neural cell adhesion molecule), and synaptosomal-associated protein 91 kDa (SNAP91), involved in cell adhesion and clathrin- dependent presynaptic vesicle assembly.
Finally, at the transcription level of the expression of the ADAMTS2 gene, antipsychotics share molecular mechanisms for the development of schizophrenia and the treatment of this disease. ADAMTS2 is localized in the brain regions like the mesolimbic and mesocortical dopamine systems. ADAMTS2 is one of the most upregulated genes in patients with schizophrenia during their first episode and is down-regulated in those who clinically improved after treatment with antipsychotics [69]. ADAMTS2 expression is activated by dopaminergic transmission (through D1-class receptors) and downstream by cAMP/CREB and mitogen-activated protein kinase (MAPK)/ERK signaling. Antipsychotic drugs inhibit D1-mediated activation of ADAMTS2 expression in neurons, which is probably involved in the clinical response to treatment in schizophrenia patients. Incubation with atypical APDs such as paliperidone, aripiprazole and risperidone leads to inhibition of ADAMTS2 basal mRNA expression with fast but transient responses (< 1 h). Clozapine and haloperidol induce a fast and sustained – up to 24 h – inhibition of ADSAMTS2 expression. These molecular effects deserve further investigation, as clozapine has superior efficacy for treatment-resistant cases as well as suicidality and ability to decrease substance abuse in schizophrenia patients [69].
POST-TRANSCRIPTION PROCESSES: MICRORNA MODULATION
MicroRNAs (miRNAs) are a class of small non-coding RNAs (ncRNAs) with relatively few nucleotides in length (22 on the average). They are transcribed from intergenic regions or spliced from introns of protein-coding genes. Next, they are transformed into mature miRNA transcript that post-transcriptionally control gene expression by binding to sites located in the 3’ UTR region of the mRNA target (3’ untranslated region), inhibiting mRNA translation. The expression and regulation of mRNA in particular have been found to be important for a wide range of biological processes and are involved in the pathophysiology of psychiatric disorders. miRNAs are stored in exosomes produced from the multi vesicular bodies of the endosomal pathway of the cell. Exosomes with miRNA are secreted from the cell of origin and can fuse with the plasma membrane of the recipient cells, unloading their miRNA cargo and post-transcriptionally modulate/inhibit multiple genes. This kind of cell-to-cell communication influences neuronal and glial development and function and, in consequence, synaptic plasticity. Post-mortem studies have shown miRNA dysregulation in the brains of patients with psychiatric disorders like schizophrenia, autism, and Tourette’s syndrome, but still little is known about the role of exosome-associated miRNA in the development of disease and its treatment [70]. Nevertheless, the central regulatory role of microRNA (miRNA) in gene expression makes them promising biomarkers. For example described changes in miRNA from people with schizophrenia include the up-regulation of hsa-miR-34a and downregulation of hsa-miR-432 [71]. However, the data for other miRNAs are conflicting. Antipsychotics may also influence the secretion of miRNA in different tissues. For example, olanzapine and haloperidol significantly reduced intracellular and exosomal miR-223 in cortical neurons but increased the secretion of miR-223 in the exosomal fraction of miRNA in astrocytes [72]. The secretion of miR-223 from astrocytes influences NMDA receptor gene expression in neurons and was detected in orbito-frontal cortex of patients who had a psychotic episode at the time of death. Increased miRNA-223 was negatively correlated with the expression of NMDA and AMPA-type subunits and GABAeric gene expression, which has clinical significance as hypofunction of NMDA transmission in the prefrontal cortex has been implicated in the cognitive and behavioral impairments in schizophrenia and other psychiatric illnesses [73]. In addition, pre-treatment with antipsychotics like haloperidol or clozapine prevented changes in miR-219 level induced by the administration of NMDA receptor antagonists (dizocilpine) in the prefrontal cortex [74]. These data support an integral role for miR-219 in the expression of behavioral aberrations associated with NMDA receptor hypofunction, involved in schizophrenia associated aberrations. Haloperidol and clozapine also appeared to regulate the expression of a large number of miRNAs related to oxidative stress and metabolism, which might be related to drug side-effects like weight gain, metabolic syndrome or insulin resistance [75, 76]. There are studies showing a decrease in plasma expression of hsa-miR-365, hsa-miR-520c-3p, and hsa-miR-181b after antipsychotic treatment, corresponding with a positive correlation between the expression of miRNA-181b and the improvement of negative symptoms [77, 78]. Finally, an in vitro antipsychotic treatment response study in T lymphocyte cell lines demonstrated that miR-200c-3p and miR-28-5p were down-regulated in haloperidol-treated cell lines in comparison to controls [79]. In contrast, the level of hsa-miR-34a and hsa-miR-432 as well as their expressions were not changed following treatment, suggesting their potential as diagnostic markers [80].
CONCLUSIONS
Despite the efficacy of antipsychotics in the treatment of schizophrenia and bipolar disorders over the last several decades their molecular mechanisms of action have turned out to be very complex and have not been fully elucidated. Recent thinking about more personalized and endophenotype-specific diagnostics and treatment requires a more advanced genomic and proteomic approach and seems to be the next step in new treatments. In conclusion, it is worth mentioning that among the proteins most expressed by antipsychotic treatment are those involved in the following: cell communication and signaling, protein metabolism, nucleic acid metabolism, transport, cell growth and maintenance, immune response, and energy metabolism. In patients who responded clinically to antipsychotic treatment the significant differences between the proteins were observed for protein metabolism pathways (twice the number of proteins in responders) and energy metabolism protein levels (shown only in non- responders). Approximately 25% of plasma proteins whose concentration significantly changed in responders was unknown and the only protein identified as increased in responders and decreased in non-responders was coronin-2A, which could be a potential biomarker [81].






