
Introduction
Wilson’s disease (WD) is a well-known metabolic disease caused by the storage and excretion of copper in the body [1]. This rare disease affects the central nervous system (CNS) and liver, resulting in a wide range of clinical findings [2]. Although the disease shows autosomal recessive and genetic transition, it is observed in 1 out of every 30,000 people in the normal population.
The ATP7B gene encodes a metal transport protein that enables the excretion of copper from the hepatocellular area. More than 300 mutations have been reported in this gene, causing cooper excretion from the liver and its accumulation in the eyes, kidneys, heart, and bones but primarily in the CNS and liver. For this reason, the disease manifests in many ways [1]. Due to the copper accumulation over time, neurological findings can occur even if the liver is not affected. Liver involvement might present itself as acute liver failure (ALF) or chronic liver disease (CLD) [3–5].
When the disease shows the first signs of ALF, the first treatment option might be liver transplantation because the condition can be urgent and life-threatening, and medical treatments might not be effective in this case [6–8]. Many patients with liver involvement other than ALF can live without symptoms if an appropriate treatment method is provided [9]. On the other hand, in patients who have not been treated properly, decompensated CLD can develop, and CNS findings are common. Thus, a liver transplant should be considered as a curative therapy in such patients [10–12].
A successfully performed liver transplant surgery can be a life-saving approach for WD [13]. At the end of first year, living donor liver transplantation provides about 85% graft and patient survival after the operation [14]. Overall transplant patients with WD show excellent overall survival and graft survival, even when compared to patients who have had transplants for other reasons. Of course, transplant patients with WD appear to be at risk for all the typical complications associated with a liver transplant [15, 16].
Aim
In this study, we aimed to assess patients with WD, who underwent a liver transplant. Furthermore, we shared the results of 14 years of experience in our transplant centre.
Material and methods
All patients who underwent a liver transplant with a diagnosis of WD in our clinic were included in the study. Ethics committee approval was obtained, and the study was conducted in accordance with the 2008 rules of the Declaration of Helsinki. All the patients who underwent liver transplant had been diagnosed according to the Leipzig criteria [4] (Table I) by gastroenterology clinics. Information was recorded based on the age, gender, weight, body-mass index (BMI), type of graft, surgical technique, and complications of the recipients and donors.
Table I
Leipzig criteria for Wilson’s disease [4]
All recipient patients were prepared according to the routine liver transplant preparation program implemented in our clinic. The tests performed included routine blood count, coagulation, comprehensive biochemistry analysis, viral serology, blood and urine culture, abdominal ultrasound-Doppler, contrast thorax-abdominal tomography, hepatic vascular reconstruction, lung function, and cardiac examination tests. All the recipients and donors – both adult and paediatric – were consulted by cardiology, infectious diseases, gastroenterology, and chest diseases departments to prove their suitability.
Donor candidates were accepted as donors if they passed the 4th-degree relative criterion or obtained ethics committee approval on the condition of blood group compatibility, which is the primary donor criterion in our clinic. Candidates ranged from 18 to 65 years old and had hepatosteatosis below 10%, no homozygous factor 5 Leiden or homozygous factor 2 mutations, and no systemic co-morbidities identified. Parenchymal structure, vascular anatomy, and biliary anatomy were visualized with magnetic resonance cholangiopancreatography (MRCP) and tri-phase computed tomography (CT) angiography. The average weight of the graft to be used tomographically in liver transplantation was calculated using the recipient’s age and weight. According to the Leipzig criteria, the 24-h amount of copper in urine and the serum ceruloplasmin values were checked for close relative donors. In the presence of doubt, the candidate was not considered as a donor and was referred to the gastroenterology clinic.
Besides the ALF, an indication of liver transplantation in CLD, high MELD score (≥ 15) or despite lower MELD score with history of oesophageal variceal bleeding or refractory ascites and persistent hepatic encephalopathy were accepted for transplant preparation.
Calcineurin inhibitors (tacrolimus or cyclosporine) and the prednisolone-based protocol were used in post-transplant immunosuppressive therapy. Mycophenolate mofetil (MMF) was added to the immunosuppression as needed. Calcineurin inhibitor doses were adjusted according to the plasma level, which was monitored daily. In the post-transplant period, the patients were followed up at least once a month in the first year, and then at an outpatient clinic every 3 to 6 months depending on the need after the second year.
Ethical statement
Ethics committee approval was obtained from the Local Ethics Committee of Sisli Memorial Hospital. The study was conducted according to the criteria of the Declaration of Helsinki and the Declaration of Istanbul. It was performed as a retrospective study with anonymized data analyses.
Results
In the years 2006–2020, 1246 liver transplants were performed in 1203 patients. The study included 27 patients with WD, including 11 females and 16 males, with a mean age of 20.8 ±11.1 years and a mean BMI of 20.5 ±3.2 kg/m2. In total, 29 liver transplants were performed on these 27 patients. The mean MELD score of the adult patients was 16.5 ±6.3, and the mean PELD score of the paediatric patients was 19.6 ±17.2.
All patients who underwent liver transplant were referred to our clinic when they were being followed up with a diagnosis of WD in various gastroenterology clinics. Five patients were referred by the intensive care units from other hospitals for ALF with grade 3–4 encephalopathy. The other 22 patients with an elective condition who had CLD were hospitalized after preparations for the liver transplant were completed.
According to the neuro-psychiatric evaluation before liver transplants, 2 patients had been using antiepileptic drugs due to epileptic diseases, and 2 patients had been using antidepressant drugs with a diagnosis of major depression. Eye examinations showed that 14 (52%) patients had Kayser-Fleischer rings. All patients except one had a ceruloplasmin level below normal.
The liver grafts comprised 5 full-size cadaveric transplants, 1 left lateral segment, 20 right lobes, and 3 left lobe transplants. The average graft weight was 833.7 ±333.8 g. The average graft/body weight ratio (GBR) was 1.6 ±0.5% in transplants. The duct-to-duct technique was performed on 22 patients, and the Roux-Y reconstruction technique was performed on 7 patients for biliary tract reconstruction.
Perioperative mortality was observed in 3 patients. All these patients had grade 3–4 encephalopathy and were accepted for urgent liver transplantation from the intensive care units of other hospitals. One of these 3 patients underwent emergency living-donor liver transplant due to primary non-function (PNF) after cadaveric donor transplant. This patient was lost due to perioperative multi-drug-resistant Klebsiella pneumoniae sepsis. The other 2 patients who underwent urgent transplants died because of brain death despite normal graft functions after liver transplantation. Even though there were 3 mortalities in 5 emergency cases, no mortality was seen among 22 patients who underwent liver transplant electively with low MELD scores.
Due to hepatic artery thrombosis (HAT) and primary non-function (PNF), 2 re-transplantations were performed. The patient who had HAT after a living-donor liver transplantation was saved with a cadaveric organ re-transplantation, and the patient is still alive. Another patient who re-transplanted due to PNF after cadaveric liver transplantation died because of sepsis after emergency re-transplantation from a live donor.
During the perioperative period, intraabdominal bleeding was observed in 2 patients. One was checked for bleeding by laparotomy, and the other was followed up with medical treatment. No further surgery was required.
Again, in the perioperative period, 6 (25%) patients had convulsion-epilepsy and were followed up with medical treatment. These patients’ antiepileptic drugs were discontinued in their follow-up during neurological monitoring. Six (25%) patients developed bile duct stenosis in the late period, and 3 patients were dilated in the biliary tract with endoscopic retrograde cholangiopancreatography (ERCP). Three patients underwent percutaneous transhepatic cholangiography (PTC) and are still being monitored.
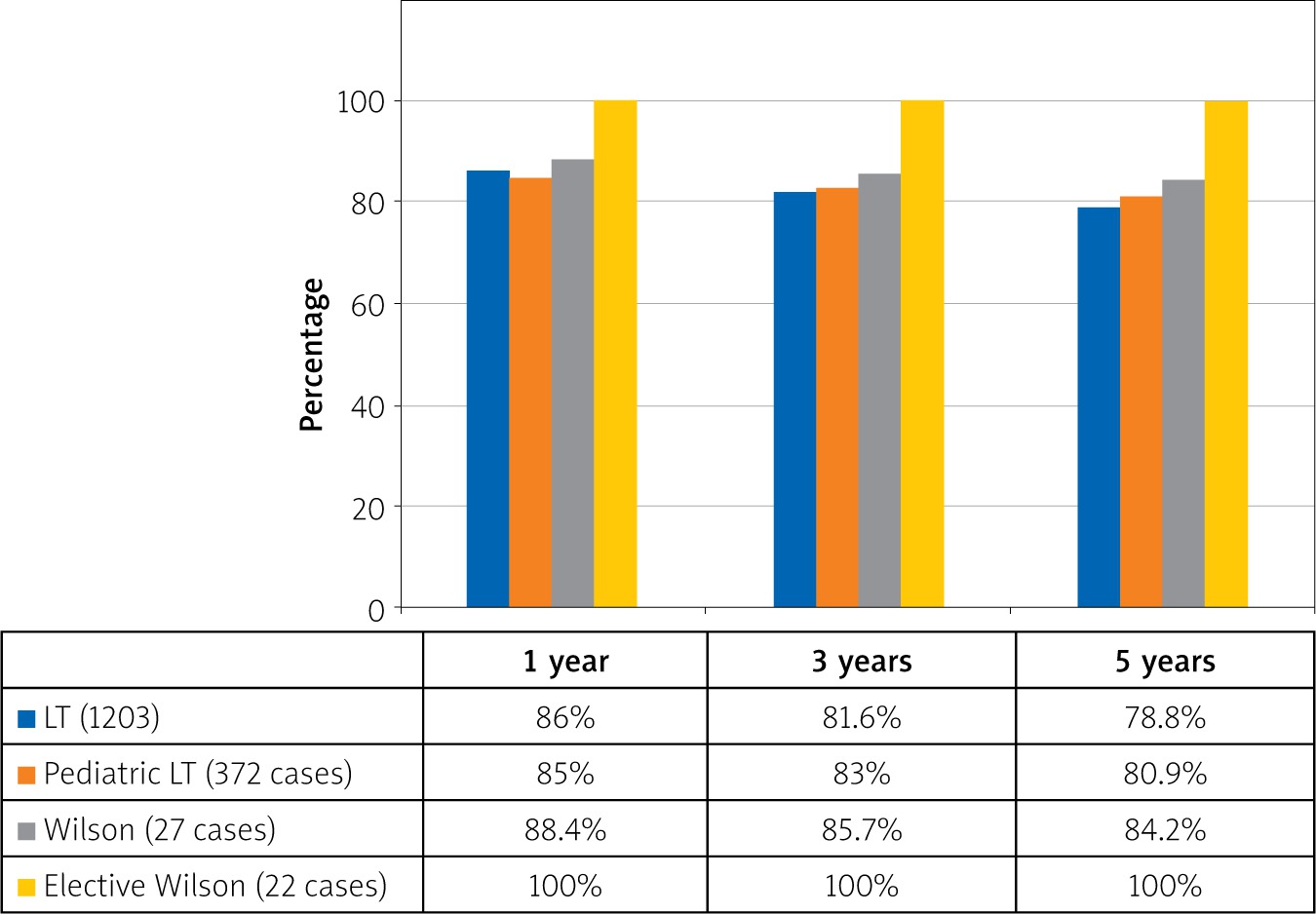
Liver recipient patients were discharged at 21.8 ±10.7 days, on average, and the mean follow-up was 6.1 ±3.7 years. There were 3 perioperative mortalities, but the other 24 patients are still alive. The overall survival of our patients was 88.8%, and the survival rates were 88.4%, 85.7%, and 84.2% among patients at 1, 3, and 5 years, respectively. The best outcome for transplant patients among the whole group was seen in elective WD patients, with a 100% survival rate in our clinic (Figure 1).
Five donor cadaveric full-size grafts were used as donors. Of the 24 live donors, 23 were 4th-degree relatives, and 1 was a more distant relative who was approved by the ethics committee. The mean age of the 9 female and 15 male live donors was 34.0 ±13.5 years, and their mean BMI was 25.1 ±4.7 kg/m2. The donors were discharged after 9.6 ±3.2 days, on average. No major complications were observed in any live donor. Two donors developed a pulmonary infection requiring the use of antibiotics, and 3 of them developed a minor wound infection that did not require any surgical intervention.
Discussion
All patients with clinical results due to a defective protein structure resulting from a mutation in the ATP7B gene are diagnosed as WD. This gene shows autosomal recessive transition and encodes a metal transport protein in the liver. As a result of this mutation, copper accumulates in the body [1]. This rare disease affects the CNS and liver in particular, and it has a wide range of clinical findings [2].
There are 2 main liver transplant indications in this disease: ALF and CLD. Recently, liver transplant has also been considered for uncontrolled neurological disease in the absence of liver deficiency [1]. Even though ALF associated with WD is the first finding in 5% of patients, its rate has been reported to be 4–6% in USA sources [17]. It was claimed that the mortality rate in these cases is 100% without a liver transplant [1, 18].
Because of the associated difficulties, the diagnosis of WD in ALF cases is still debatable. The most important diagnostic tools include 24-h urine levels of copper, hepatic copper concentration, ATP7B gene mutation, Kayser-Fleischer rings, neurological symptoms, cranial MRI imaging, serum ceruloplasmin and copper levels, and haemolytic anaemia. However, Kayser-Fleischer rings are only present in 50% of patients [19], as is the case in our patients with WD. Serum and urine copper are unreliable parameters because they change not only in WD but also in other patients with ALF [20, 21]. These situations make the diagnosis difficult, and the differential diagnosis is crucial.
Five of the WD with ALF liver transplant patients, performed in our clinic, had been diagnosed by various gastroenterology clinics. Three of these 5 patients had a cadaveric graft with an emergency call, and 2 had a live donor graft. All patients were intubated with grade 3-4 encephalopathy when admitted from intensive care units. Mortality in liver transplantation due to WD has been observed only in this patient group. In 2 patients, brain death occurred, but the graft functions were normal after cadaveric liver transplantation. For the other lost patient, a living-donor liver transplant was performed with a diagnosis of PNF after cadaveric liver transplant. The patient died due to multi-drug-resistant Klebsiella pneumoniae sepsis in the perioperative period.
Supportive treatments are used to save time in patients referred with a diagnosis of ALF, especially in the presence of deep encephalopathy, during live-donor preparation or during the waiting time for a cadaveric donor. Examples include exchange transfusion, plasmapheresis, the molecular adsorbent recycling system (MARS), fractionated plasma separation and absorption (FPSA), and kidney replacement treatments [22, 23]. In addition to MARS, FPSA, and kidney replacement treatments, a healing effect has been demonstrated for plasma exchange treatments, especially in serum copper, liver function, and kidney function [24]. As a clinical protocol, we perform plasmapheresis as a bridge therapy for all patients who are taken to intensive care because of ALF.
There has been little discussion on liver transplant decisions in CLD cases related to WD. The issue that can be discussed is that in countries where the operations performed are mainly cadaveric, liver transplantation is based on the MELD score and patients only have the chance of transplantation when the MELD score rises. On the other hand, a living-donor liver transplantation can give a chance to WD patients with CLD in better clinical status by preparing appropriate donors under elective conditions. In our series, 22 patients with low MELD score underwent elective liver transplants, and 21 of them were from live donors. All the patients who underwent elective surgeries are still alive. Moreover, our study shows that the best outcome among the liver transplant patients belonged to transplanted patients due to WD with CLD (Figure 1).
Recently, there have been reports of cases with CNS involvement that do not have liver failure as a third liver transplant indication in WD [25]. The issue of liver transplant and CNS deformation is debatable, especially in patients with advanced neurological involvement. However, it has been reported that liver transplant improves neurological involvement or prevents the disease from getting worse [18, 26]. Survival is poor in CLD patients with severe neurological involvement [1, 18]. On the other hand, as well as the risk of mortality and morbidity of the liver transplant operation, using lifelong immunosuppressant drugs also comes with its own challenges for non-CLD patients. Furthermore, there can be potential donor complications, especially in countries that use live donors. Therefore, when the possible inconveniences of donors and recipients who do not suffer from liver function deficiency are taken into account, careful patient selection becomes essential.
All our cases had liver transplants due to liver failure, and no liver transplant was performed due to only neurological disease without CLD. Non seizures were observed in the patients who had neurological symptoms preoperatively and postoperatively and whose antiepileptic and antidepressant drugs were stopped in their neurological follow-up after the liver transplantation.
Conclusions
Liver transplant is a successful treatment method with a high survival rate in the treatment of WD-related, acute or chronic liver failure. Living-donor liver transplant has an important advantage for transplant patients with CLD before the liver disease progresses or in ALF cases, before prolongation of the organ waiting time.











