
Background
The lysosomal storage diseases are a group of genetic disorders resulting from defective lysosomal metabolism [1]. Most lysosomal storage diseases are characterized by a large phenotypic spectrum of unspecific disease manifestations, leading to considerable diagnostic problems. Among these disorders should be mentioned mucopolysaccharidoses, mucolipidoses, glycoprotein storage disease, and others. The enzyme deficiencies have an autosomal recessive basis, with the exception of Hunter mucopolysaccharidosis II, which is X-linked recessive, and Fabry disease, which is X-linked with frequent manifestations in females. The target organs are determined by the usual sites of degradation for a macromolecule. All the disorders are progressive, and many are fatal in childhood or adolescence [2]. Diagnosis of lysosomal storage diseases is difficult because there are a number of overlapping clinical features that are not specific for lysosomal storage diseases [3, 4].
There is extensive clinical, biochemical, and molecular heterogeneity within the lysosomal storage diseases. In most instances, different mutations within the structural genes for lysosomal enzymes account for varying degrees for severity from individual to individual and for the diverse combinations of visceral, skeletal, neurological, ocular, and other manifestations [5].
There are limited data on the incidence of lysosomal storage diseases, probably because of the rarity and heterogeneity of the condition [6–10]. Newborn screening pilot studies reported on average a 5–80 times higher birth prevalence than previously reported [11–14].
The reported data address the incidence of lysosomal storage diseases, obtained from the public health service databases in Poland. The data are given by subtypes from the National Health Fund from 2013 through 2015.
Material and methods
Source of data
Lysosomal storage disease patients were identified from the Polish National Health Fund database (2013–2015). In order to ensure that the reported incidence data included newly hospitalized patients, anyone previously discharged with a lysosomal storage disease diagnosis was excluded.
Outcome variables
The 10th revision of the International Classification of Diseases (ICD-10) was used to identify all first hospital admissions for the outcome variables amyloidosis E75: E75.0 GM2 gangliosidosis, E75.1 other gangliosidosis, E75.2 other sphingolipidosis, E75.3 sphingolipidosis, unspecified, E75.4 neuronal ceroid lipofuscinosis, E75.5 other lipid storage disorders, and E75.6 lipid storage disorder. However, because diagnostics is time consuming, the initial discharge may be changed. Therefore, we used the discharge diagnosis of the last hospitalization. Diagnostic details are not included among the discharge data.
All individual identified entities of lysomal storage disease group belong to disorders of sphingolipid metabolism and other lipid storage disorders (ICD10: E75). However, we excluded it because of many reporting errors and trying to avoid homogeneity of the group by this one disease. In pre-analysis E75 comprises nearly 40% of all cases on the lysomal storage disease group, which disrupted the study.
Individual variables
Children, adults, males, and females were included in the study. The geographic region of residence in 2013–2015 was divided into 6 regions: Masovia and Silesia with the largest population – each of them about 5.0 million habitants), and 4 others regions (south-east, south-west, north-east, and north-west). Thus, all these regions were comparable in terms of number of habitants.
Statistical analysis
The results were calculated to provide information on the incidence rate of rare metabolic diseases in lysosomal storage diseases in Poland according to the region of residence where it occurs, age, gender, and subtypes of diagnosis. Person-years were calculated from the start of follow-up on 1 January 2013 until hospitalization for lysosomal storage diseases, death, or the closing date (31 December 2015). We used R v. 3.6.1 for the statistical analyses. Charts included in the publication were created by using the R data visualization library – ggplot2 version 2.1.0 and Microsoft Excel 2013.
Results
Case numbers and incidence of hospitalized lysosomal storage disease patients are shown in Table I. For the years 2013 to 2015, a total of 212 patients were identified, giving an incidence of 1.84 per million person-years. Other sphingolipidosis (E75.2) was the largest disease category with 127 patients (incidence 1.1 patients/million habitants), followed by GM2 gangliosidosis (E75.0) with 29 patients (incidence 0.25 patients/million habitants).
Table I
Numbers and incidence rate (per million person-years) for hospitalized lysosomal storage diseases patients, 2013-2015
Table II presents the incidence rate of lysosomal storage diseases for men and women. Overall, men had a higher incidence than women (respectively, IR = 2.53 and IR = 1.84).
Table II
Incidence rate of lysosomal storage diseases for men and women, 2013-2015
The age-specific rate of hospitalization for lysosomal storage diseases is shown in Figure 1A–G1.
Regional differences in the incidence of lysosomal storage diseases are shown in Table III.
Figure 1
Age-specific incidence rate (per million person-years) of hospitalized patients with lysosomal storage diseases by subtypes, 2009–2015: A) E75.0 GM2 gangliosidosis; B) E75.1 Other gangliosidosis; C) E75.2 Other sphingolipidosis; D) E75.3 Sphingolipidosis, unspecified; E) E75.4 Neuronal ceroid lipofuscinosis; F) E75.5 Other lipid storage disorders; Age-specific incidence rate (per million person-years) of hospitalized patients with lysosomal storage diseases by subtypes, 2009–2015: G) E75.6 Lipid storage disorder, unspecified
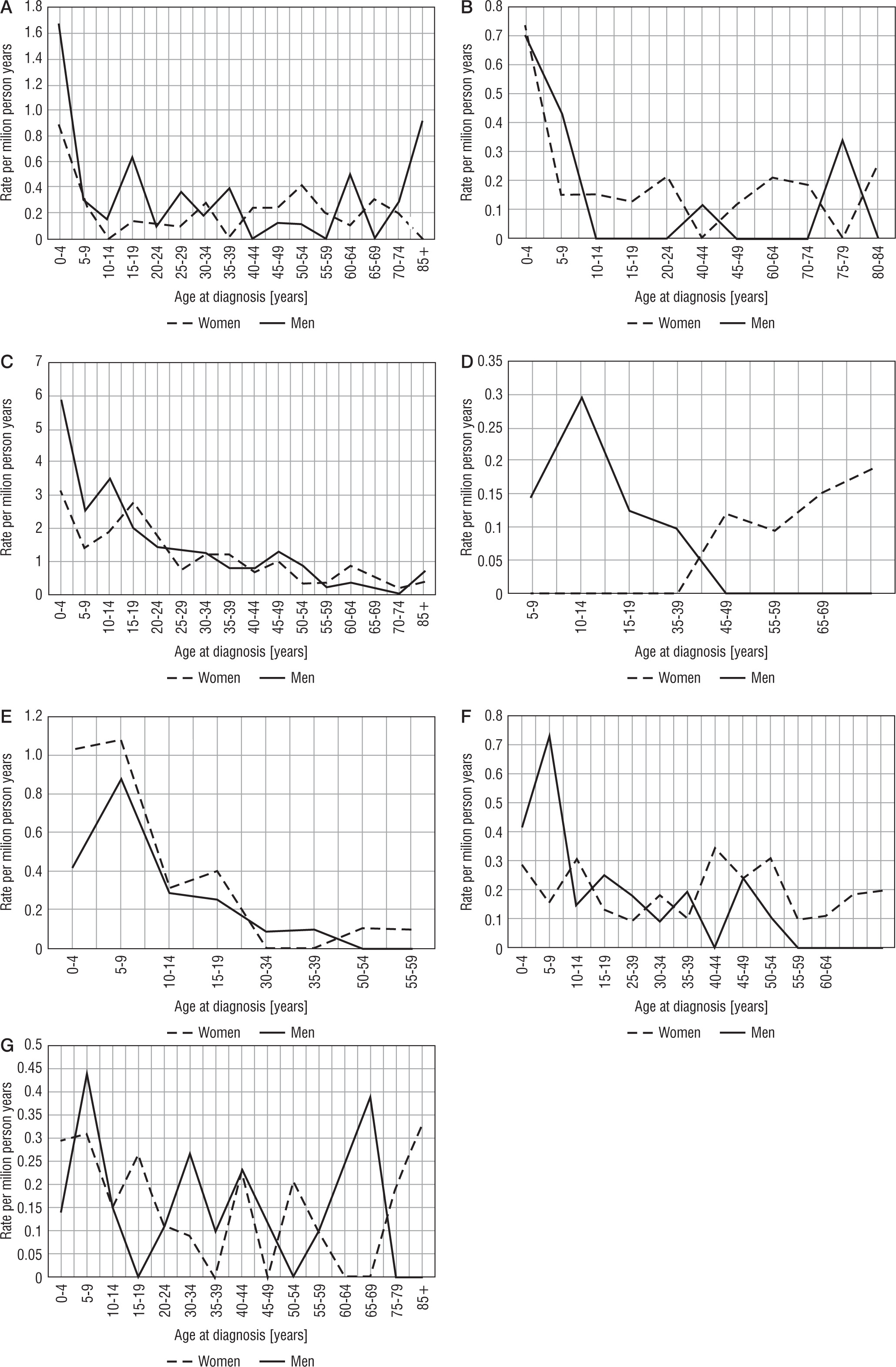
Table III
Regional differences in the incidence of lysosomal storage diseases
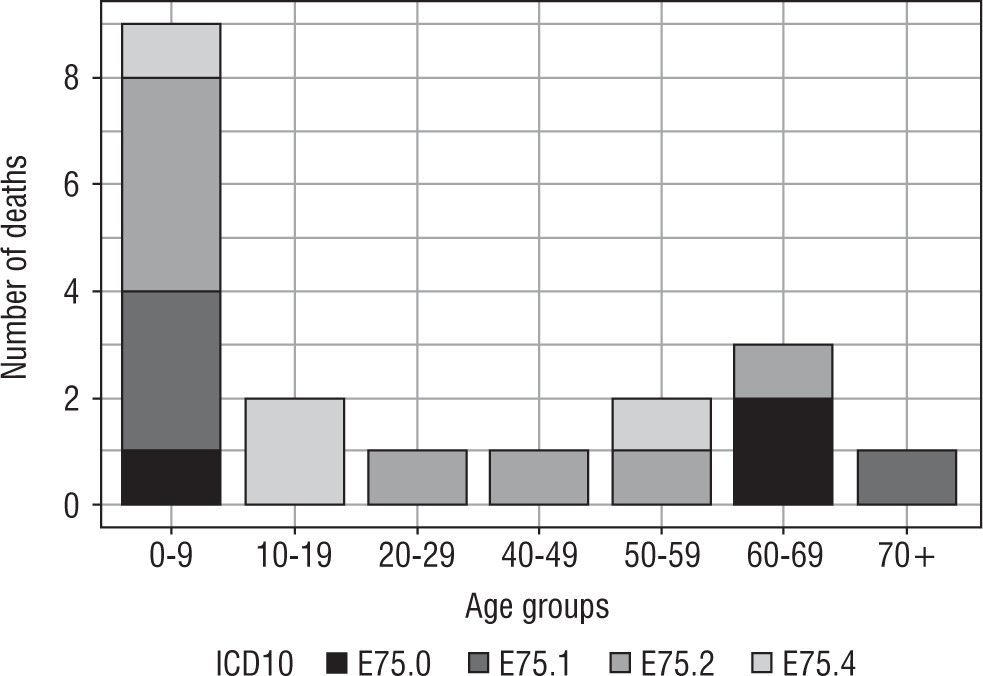
Number of deaths among patients with lysosomal storage diseases hospitalized between the years 2013 and 2015 are presented in Figure 2.
Discussion
The reported study provides comprehensive information on lysosomal storage diseases in the Polish population, which is approximately 38 million people, and therefore the acquired data on lysosomal storage diseases incidence are very accurate. Our research demonstrated that during the years 2013 to 2015, 212 patients with lysosomal storage diseases were hospitalized (incidence of 1.80 per million person-years).
In order to obtain reliable data in our study, we took into account the medical records of all the patients who had received services of the public healthcare system in Poland from 2013 to 2015, using National Health Found data. Data of almost all the people of Polish nationality, hospitalized in Poland, are uploaded into the National Health Found database. The National Health Found is the payer for medical services received by all insured subjects.
Only scarce epidemiological data for lysosomal storage diseases were published in the past [2].
As indicated in our paper, the number of patients hospitalized in Poland in 2013-2015 due to lysosomal storage diseases was 212 (incidence rate 1.84 patients/million habitants * year). The most common reason for hospitalization due to the above-mentioned conditions was so-called other sphingolipidosis (incidence rate 1.1 patients/million habitants * year). The next most common reason for the frequency of hospitalization due to lysosomal storage diseases were GM2 gangliosidosis sphingolipidosis (incidence rate 0.25 patients/million habitants * year) (Table I).
The number of data on the incidence of lysosomal storage diseases is very limited. In the work of Kingma et al. [2] the data from the literature on the above-mentioned topic are summarized. However, in the analysed studies, there was no necessity for hospitalization, but only birth prevalence of lysosomal storage diseases in Australia, the Netherlands, British Columbia, Portugal, the Czech Republic, and the United Arab Emirates [6–8, 10, 15]. This disease ranged from 7.5/100,000 births in British Columbia to 23.5/100,000 births in the United Arab Emirates [10, 15]. These data are difficult to compare with the data obtained by us. As can be seen from the above that the risk of birth prevalences of lysosomal storage diseases in the above-mentioned countries was significantly higher than in Polish conditions. However, it should be admitted that the cited studies considered the birth prevalence of lysosomal storage diseases, whereas in our work we examined only the patients hospitalized due to lysosomal storage diseases. This could be one of the main reasons for the differences in the obtained test results. We believe, however, that this does not explain the fully proven differences. We are of the opinion that the differences could also result from the fact that Poland is a country in which only people of Polish origin live in the vast majority, and hence the frequency of mutations in the genes responsible for the occurrence of lysosomal storage diseases can be relatively small, whereas in people of other origin (unlike other analysed countries) [7, 8, 10, 15] this frequency is significantly higher. The reason for the differences may also be an imperfect system of recognizing these diseases in our country. Screening programs for lysosomal storage diseases may prevent irreversible organ damage. Poland lacks this program. However, interestingly, as in our observation, the most common lysosomal storage diseases were sphingolipidosis, followed by gangliosidosis (Table I) [2].
The incidence of hospitalization due to lysosomal storage diseases was significantly higher in men (IR 2.15, 95% CI: 1.76–2.53) than in women (IR = 1.84, 95% CI: 1.59–2.0) (Table II). In the available literature, we were unable to find work on differences in morbidity due to lysosomal storage diseases between women and men (Table II). Therefore, no justification for these differences was found. Perhaps some of these conditions also depend on changes in the X chromosome. X-linked recessive with incomplete penetrance in heterozygous females. The condition affects hemizygous males (i.e. all males), as well as homozygous, and in many cases heterozygous females. While males typically experience severe symptoms, women can range from being asymptomatic to having severe symptoms. New research suggests that many women suffer from hypertrophic left ventricular heart problems and kidney failure. These X-inactivation patterns during embryonic development of the female chromosome X occur only 1 time in men. In this situation, it is more likely to reveal recessive changes characteristic, for example, for Fabry’s disease.
Hospitalizations due to lysosomal storage diseases were most common in young people (up to 24 years of age) with the exception of hospitalization caused by lipid storage disease-unspecified (Fig. 1). Lysosomal storage diseases occur at a young age; hence, the fact that the necessity of hospitalization takes place at a young age is beyond doubt. Due to poor prognosis in patients with lysosomal storage diseases, the incidence of hospitalization at a later age is less frequent (Fig. 1). This is consistent with the data presented in Figure 2. In turn, the highest mortality rate (as expected) was demonstrated in people aged 0–9 years (Table II).
The study also analysed the frequency of hospitalization due to lysosomal storage diseases in various regions of our country (Table III). Because the number of hospitalizations, in general, was small due to the rarity of these diseases, there were no significant differences between individual regions of Poland (Table III). The number of patients hospitalized in Poland due to lysosomal storage diseases in the years 2013–2015 was relatively small. After dividing into individual regions, we had to deal only with individual cases in individual parts of the country. For this reason, the analysis did not bring new cognitive elements.
Study limitations
Certain errors may be suspected in our analysis.
First, it is not absolutely certain that all the cases of lysosomal storage diseases belonged to that group of patients. A more precise diagnostic analysis would have qualified the cases to another group.
Second, we may suspect that some cases of lysosomal storage diseases, especially in patients of health centres in smaller towns or in rural environments, were not properly diagnosed. This is perhaps the reason for the significantly lower incidence of lysosomal storage diseases in the general Polish population, when compared to other populations.
Nonetheless, we present, for the first time, complete and reliable data on the incidence rate of lysosomal storage diseases in Poland. This may help better understand this issue, providing improved diagnostics of this specific condition and, consequently, ensuring more targeted treatment.
Conclusions
The incidence rate of lysosomal storage diseases in Poland is about 1.84 per million person-years. Other sphingolipidosis (E75.2) was the largest disease category, with 127 patients (incidence 1.1 patients/milion habitants), followed by GM2 gangliosidosis (E75.0), with 29 patients (incidence 0.25 patients/million habitants). The hospitalization rate of lysosomal storage diseases was higher in mar than women . No statistically significant differences were observed among the recorded incidence rates obtained at the analysed Polish regions.









