18.06.2022
Redaktor:
Krystian Lurka
Źródło: Psychiatria Spersponalizowana/Marek Krzystanek, Robert Krysiak, Adam Chabrzyk i Artur Pałasz
Farmakologiczne możliwości leczenia zaburzeń libido

Zaburzenia seksualne związane z cyklem reakcji seksualnej są częstym problemem w populacji ogólnej – przyczyny zaburzeń seksualnych mogą być różne i złożone, podobnie jak ich leczenie, które w każdym przypadku wymaga personalizacji, a w sytuacji braku poprawy – podejmowania kolejnych prób z wykorzystaniem danych pochodzących z publikacji klinicznych.
Artykuł Marka Krzystanka z Kliniki Rehabilitacji Psychiatrycznej Wydziału Nauk Medycznych Śląskiego Uniwersytetu Medycznego w Katowicach, Roberta Krysiaka z Kliniki Chorób Wewnętrznych i Farmakologii Klinicznej Wydziału Nauk Medycznych Śląskiego Uniwersytetu Medycznego w Katowicach, Adama Chabrzyka z Oddziału Psychiatrycznego w Nowym Szpitalu w Olkuszu, Artura Pałasza z Zakładu Histologii Wydziału Nauk Medycznych Śląskiego Uniwersytetu Medycznego w Katowicach:
Podstawą prawidłowych funkcji seksualnych jest brak patologii w budowie i funkcjonowaniu części układu nerwowego związanych z reakcją seksualną i w narządach płciowych, prawidłowa czynność gonad z prawidłowym stężeniem estrogenów, progesteronu i testosteronu oraz brak zaburzeń somatycznych, psychologicznych i psychiatrycznych, mogących zakłócać funkcje seksualne, albo jak w przypadku problemów medycznych – konieczność stosowania leków z objawami niepożądanymi w postaci zaburzeń seksualnych. W leczeniu zaburzeń seksualnych możliwe są różne strategie, obejmujące psychoterapię, farmakoterapię/hormonoterapię, modyfikację leczenia farmakologicznego powodującego objawy niepożądane w postaci dysfunkcji seksualnych lub metody zabiegowe. Autorzy dokonali przeglądu badań klinicznych, poszukując danych dotyczących farmakologicznego leczenia zaburzeń pożądania. W aspekcie farmakologicznego leczenia zaburzeń pożądania większość badań ma charakter opisów przypadków, serii przypadków i badań przeprowadzonych na małych grupach osób. Farmakologiczne leczenie zaburzeń pożądania powinno być zarezerwowane do leczenia pierwotnych zaburzeń seksualnych albo wspomagającego leczenia psychogennych i somatogennych zaburzeń. Leczenie w każdym przypadku wymaga personalizacji, a w sytuacji braku poprawy podejmowania kolejnych prób z wykorzystaniem dostępnych danych z publikacji klinicznych.
Wstęp
Współczesnej seksuologii udało się opisać fizjologiczny, liniowy wzorzec reakcji seksualnej człowieka, będący podstawą do opisu nieprawidłowości pojawiających się w przebiegu stosunku. Według tego modelu reakcja seksualna przebiega u kobiet i mężczyzn bez zaburzeń seksualnych przez kolejne fazy pożądania, podniecenia, plateau, orgazmu i odprężenia. Linearny model reakcji seksualnej nie został dotąd sfalsyfikowany i jest nadal podstawą do opisywania zaburzeń seksualnych człowieka, jako odchyleń od tej fizjologii 1, 2.
W linearnym modelu zaburzeń seksualnych kluczowe wydaje się pożądanie seksualne. W odniesieniu do pożądania seksualnego, obecnie rozróżnia się dwa jego rodzaje: spontaniczne i reaktywne, przy czym pożądanie u mężczyzn może nie występować albo jest tożsame z uświadamianym podnieceniem seksualnym 3. Brak pożądania (lub pożądania utożsamianego z podnieceniem) uniemożliwia fizyczną reakcję podniecenia i odbycie stosunku seksualnego.
Przyczyny zaburzeń pożądania są złożone i mogą mieć charakter psychologiczny i/lub somatyczny. W leczeniu tych zaburzeń możliwe są różne strategie, obejmujące psychoterapię, farmakoterapię/hormonoterapię, modyfikację leczenia powodującego objawy niepożądane w postaci dysfunkcji seksualnych lub metody zabiegowe.
Celem niniejszego artykułu jest z jednej strony przedstawienie biologicznych uwarunkowań pożądania, a z drugiej – omówienie dostępnych doniesień na temat możliwości farmakologicznej korekcji zaburzeń libido. Informacje te mogą mieć praktyczne znaczenie dla lekarzy szukających farmakologicznych sposobów poprawy libido u kobiet i mężczyzn.
Biologiczne uwarunkowania libido
Libido jest złożoną funkcją seksualną, na którą wpływają czynniki psychologiczne i biologiczne. Neurofizjologiczne tło zaburzeń pożądania stanowią przede wszystkim dysfunkcje układu nagrody, a w szczególności budujących go struktur układu mezolimbicznego. Wchodzące w jego skład szlaki regulatorowe kontrolowane są przez neurony układu serotoninergicznego i noradrenergicznego, syntezowane miejscowo i obwodowo hormony neuropeptydowe, u kobiet przez estrogeny i progesteron, u mężczyzn natomiast przez poziom testosteronu. Liczne badania podstawowe oparte na modelach zwierzęcych oraz obserwacje kliniczne umożliwiły identyfikację endogennych czynników pobudzających i hamujących reakcje seksualne (rycina 1).
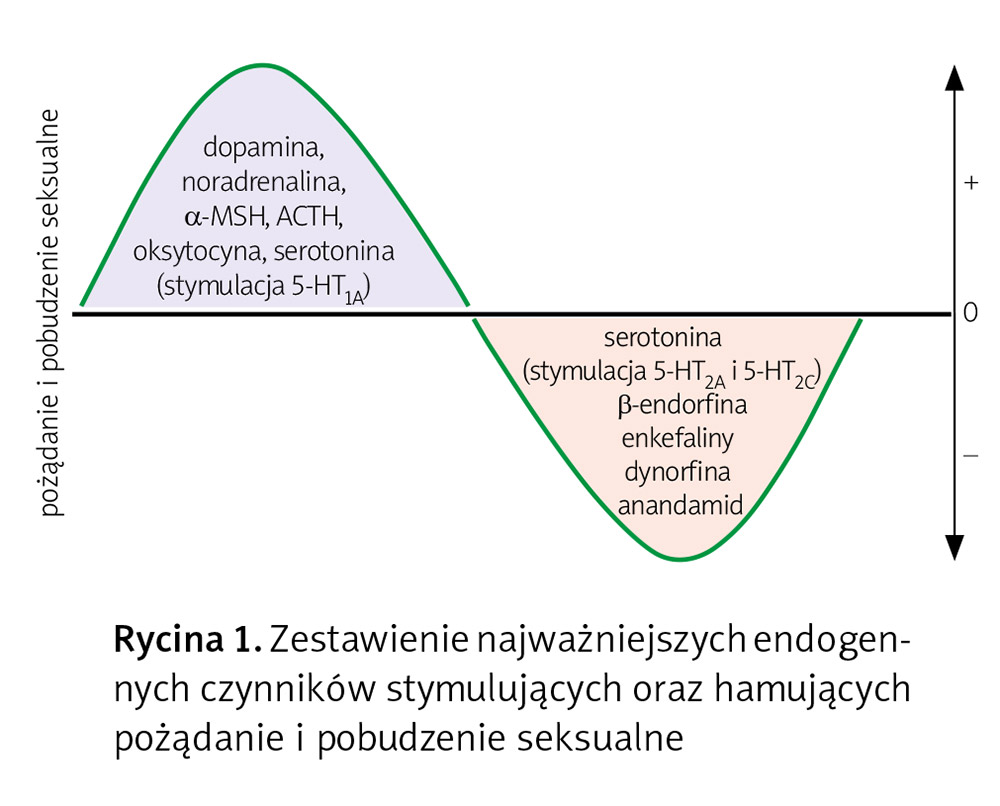
Inicjujące stan pożądania neurony dopaminergiczne systemu mezolimbicznego zlokalizowane są w polu brzusznym nakrywki (VTA), skąd oddają swe projekcje do szeregu struktur podkorowych, w szczególności do ciała migdałowatego i jądra półleżącego. Istotną rolę w generowaniu reakcji seksualnych przypisuje się również neuronom dopaminergicznym warstwy niepewnej (zona incerta), których aksony docierają do przyśrodkowego pola przedwzrokowego podwzgórza (mPOA). Efektem selektywnego zniszczenia zarówno mPOA, jak i jądra półleżącego w mózgu szczura jest daleko idąca redukcja zachowań seksualnych 4. Rola dopaminy w inicjowaniu pobudzenia seksualnego leży u podstaw wpływu leków neuropsychiatrycznych na stan pożądania u ludzi. Przykładowo L-DOPA (3,4-dihydroksy-L-fenyloalanina) wykazuje działanie stymulujące reakcje seksualne, które może być wygaszane działaniem nieselektywnych antagonistów receptorów dopaminergicznych. W związku z tym współcześnie stosowane leki antypsychotyczne, będące w większości antagonistami transmisji dopaminowej, mogą zaburzać szeroko rozumianą aktywność seksualną obserwowaną zarówno w modelach zwierzęcych, jak i w badaniach klinicznych. Niewykluczone, że kluczową rolę w dopaminergicznej stymulacji seksualnej u zwierząt może odgrywać ilościowy stosunek receptorów D1 względem D2 w obszarze mPOA. Zgodnie z tym modelem, aktywacja receptora D1 wyzwala psychiczne składowe pożądania, natomiast stymulacja receptora D2 związana jest raczej czynnością wykonawczą układu płciowego 5. Szlaki noradrenergiczne mózgowia, których głównym źródłem są neurony miejsca sinawego (locus coeruleus) pnia mózgu, inicjują pobudzenie seksualne i podtrzymują je drogą stymulacji układu współczulnego. Oddają one liczne projekcje zaopatrujące podwzgórze, układ limbiczny i pewne okolice kory nowej. Agoniści receptora α2, jak klonidyna, zmniejszają uwalnianie neurotransmitera, czego efektem jest depresja układu współczulnego i obniżenie wrażliwości na bodźce o charakterze erotycznym 6, antagoniści natomiast, m.in. johimbina, wywołują efekt przeciwny, dlatego bywają niekiedy stosowane jako stymulanty seksualne. Inhibitory syntezy noradrenaliny, np. dietyloditiokarbaminian, wyciszają przejawy podniecenia seksualnego u zwierząt. Neurohormonem włączonym w ośrodkowe mechanizmy generowania podniecenia seksualnego jest również oksytocyna, nonapeptyd syntezowany w wielkokomórkowych jądrach neurosekrecyjnych podwzgórza. Iniekcja oksytocyny do mPOA lub jądra brzuszno-przyśrodkowego podwzgórza (VMH) szczurów powoduje w przypadku samic wzmożenie behawioralnych efektów pobudzenia seksualnego (lordoza), natomiast u samców stymuluje erekcję 7. Stymulacja receptorów dopaminergicznych D1 w obrębie jądra przykomorowego podwzgórza (PVN) powoduje zarówno uwolnienie oksytocyny, jak i wzrost poziomu dopaminy w jądrze półleżącym, co sugeruje obecność zależnego od oksytocyny systemu integrującego podwzgórzowy i mezolimbiczny szlak dopaminergiczny 8. Kolejnymi neuropeptydami wyzwalającymi stan pożądania i pobudzenia seksualnego są dwie pochodne proopiomelanokortyny (POMC): adrenokortykotropina (ACTH) oraz melanotropina (α-MSH), których cząsteczki wiążą się ze swoistymi receptorami melanokortynowymi MC3 i MC4 licznych neuronów podwzgórza i układu limbicznego 9. Źródłem melanokortyn są neurony jądra łukowatego podwzgórza, oddające liczne rozproszone projekcje zarówno do struktur sąsiadujących, jak i odległych, w tym układu limbicznego, śródmózgowia i pnia mózgu. Neurony te wykazują wrażliwość na steroidy, zaobserwowano bowiem podwyższenie poziomu α-MSH w podwzgórzu samic szczurzych po podaniu estradiolu 10. Farmakomodulacja receptorów melanokortynowych (MCRs) wydaje się niezwykle obiecującą strategią terapeutyczną w zaburzeniach seksualnych.
Kluczową rolę w hamowaniu stanu pożądania odgrywają szlaki serotoninergiczne biorące swój początek w śródmózgowiowych jądrach szwu (nuclei raphes). Aksony opuszczające tę strukturę docierają do licznych okolic ośrodkowego układu nerwowego (OUN): kresomózgowia, podwzgórza, układu limbicznego, hipokampa, pnia mózgu oraz eferentnie do rdzenia kręgowego, gdzie zaopatrują autonomiczne ośrodki w lędźwiowej i krzyżowej części rdzenia kręgowego, kontrolujące odruchy genitalne. Co warte podkreślenia, serotonina hamuje funkcje seksualne drogą stymulacji receptorów 5-HT2A i 5-HT2C, efektem aktywacji autoreceptorów 5-HT1A jest natomiast wzrost pożądania i redukcja uwalniania neurotransmitera do szczeliny synaptycznej 11. Neurotoksyczne uszkodzenie zstępujących dróg serotoninergicznych rdzenia kręgowego silnie pobudza erekcję, co sugeruje, że odruch wzwodu jest stale tonicznie hamowany przez te szlaki 12. Istotne opóźnienie ejakulacji oraz anorgazmia to efekty bardzo często obserwowane u pacjentów przyjmujących leki przeciwdepresyjne z grupy selektywnych inhibitorów wychwytu zwrotnego serotoniny (SSRIs). Zjawiska te mogą być, przynajmniej częściowo, odwrócone przez podanie oksytocyny. Szczury poddane działaniu DOI (2,5-dimetoksy-4-jodoamfetamina), antagonisty receptorów 5-HT1C i 5-HT2, manifestowały zahamowaną aktywność seksualną 13. Zastosowanie psychoaktywnego agonisty receptorów 5-HT1C i 5-HT2 – TFMPP (3-trifluorometylofenylopiperazyna) – powodowało zniesienie odruchów kopulacyjnych u samców królików. Ważnymi inhibitorami pobudzenia seksualnego są endogenne peptydy opioidowe, zarówno pochodne POMC, takie jak β-endorfna (β-END), jak i proenkefaliny (Met- i Leu- enkefalina) oraz prodynorfiny (dynorfiny A i B), manifestujące zróżnicowane powinowactwo do receptorów opioidowych μ, δ i κ. Głównym źródłem opioidów są neurony POMC jądra łukowatego podwzgórza wysyłające liczne projekcje do kresomózgowia, śródmózgowia, pola brzusznego nakrywki, jąder półleżących i prążkowia. Podanie agonistów receptora opioidowego μ do przyśrodkowego mPOA lub VMH podwzgórza hamuje zachowania seksualne szczurów 14. Prawdopodobnie uwolnienie opioidów w mPOA aktywuje układ nagrody i jest czynnikiem generującym stan refrakcji seksualnej 15. Z odmienną sytuacją mamy do czynienia w przypadku VTA, gdzie uwolnienie opioidów pociąga za sobą stymulację aktywności seksualnej, a celowana infuzja morfiny lub dynorfiny do tej okolicy wyzwala szereg behawioralnych symptomów pobudzenia seksualnego u szczurów obydwu płci 16. Dokomorowa iniekcja agonistów receptora δ skutkuje uaktywnieniem szeregu zachowań seksualnych, natomiast iniekcja do mPOA wywołuje efekt przeciwny 17. Substancjami wygaszającymi aktywność seksualną są również endogenne kanabinoidy, m.in. anandamid (arachidonyloetanoloamina, AEA) czy 2-arachidonyloglicerol (2-AG), działające za pośrednictwem receptora kanabinoidowego typu 1 (CB1) obecnego w licznych strukturach podwzgórza i układu limbicznego. Najwyższe stężenie endogennych kanabinoidów w podwzgórzu stwierdzono u samic szczura w okresie międzyrujowym (diestrus), kiedy aktywność seksualna zwierząt jest istotnie obniżona, po czym w miarę przechodzenia w kolejne fazy cyklu rozrodczego następował jego stopniowy spadek 18. Hormonem hamującym reakcje seksualne, w tym stany pożądania i ekscytacji, jest również prolaktyna, peptyd produkowany przez komórki kwasochłonne przysadki gruczołowej, odgrywający wiodącą rolę w regulacji cyklu jajnikowego i procesu laktacji. Podstawowym inhibitorem sekrecji prolaktyny jest dopamina. Blokada transmisji dopaminergicznej prowadzi zatem do hiperprolaktynemii, której efektem jest stymulacja hamującej sygnalizacji GABA-ergicznej i opioidowej, co istotnie obniża poziom libido. Najnowsze badania sugerują również wpływ endogennych steroidów o potencjalnych cechach ludzkich feromonów płciowych: androsta-4,16,-dien-3-onu oraz estra-1,3,5(10),16-tetraen-3-olu na podwzgórzowe mechanizmy pożądania i podniecenia seksualnego. Ich efekty fizjologiczne wydają się zależne od płci i orientacji seksualnej 19. Podwzgórzowo-limbiczne mechanizmy pożądania podlegają pewnym odmiennościom ze względu na płeć. Na rycinie 2 przedstawiono przykładowo regulację pożądania u kobiet.

Farmakoterapia zaburzeń libido
Zaburzenia libido mogą mieć charakter pierwotny albo wtórny, spowodowany np. zażywanymi lekami albo zaburzeniami hormonalnymi. W sytuacji potwierdzonych zaburzeń poziomu hormonów płciowych we krwi, w leczeniu zaburzeń libido stosuje się miejscową lub systemową substytucję hormonalną estrogenami, substytucję systemową testosteronem lub progesteronami albo podawanie selektywnego modulatora receptora estrogenowego – tibolonu lub ospemifenu.
Hormonalna terapią zastępcza (HTZ) ma mały do średniego wpływ na poprawę pożądania i podniecenia u kobiet w okresie około- i pomenopauzalnym. Odgrywa natomiast ważną rolą w przywróceniu troficzności nabłonka pochwy, likwidując nieprzyjemne odczucia przy stosunku czy dyspareunię 21.
W leczeniu zaburzeń libido u kobiet poprawę może przynieść podawanie androgenów, zwłaszcza w przypadku małego stężenia całkowitego testosteronu i siarczanu dehydroepiandrosteronu. Jeszcze szerzej wykorzystuje się leczenie za pomocą androgenów zaburzeń pożądania u mężczyzn, pod warunkiem że ich stężenie mieści poniżej normy dla wieku. W większości krajów europejskich i w USA dostępne są różne formy podawania testosteronu (tabletki, iniekcje domięśniowe, kremy, żele, plastry, tabletki dopoliczkowe, aerozol do aplikacji pod pachę), przy czym stosowane dawki są znacznie większe niż dla kobiet, co odzwierciedla fizjologiczne różnice w dobowym wytwarzaniu tego hormonu (około 5–10 mg u mężczyzn i 150–300 μg u kobiet) 21, 22. Natomiast egzogenny dehydroepiandrosteron (prasteron) jest stosowany w zbliżonych dawkach u obu płci, przy czym w leczeniu zmniejszonego libido preferuje się doustną drogę podaży leku.
Substytucja testosteronu u mężczyzn może poprawiać zaburzenia pożądania, jednak – jak wspomniano – jest ona skuteczna praktycznie wyłącznie w przypadkach potwierdzonego zmniejszenia stężenia tego hormonu w osoczu 22. Podawanie testosteronu kobietom, zwykle w postaci plastrów uwalniających 300 μg hormonu na dobę (dawka umożliwiająca uzyskanie stężenia testosteronu w górnych granicach normy dla kobiet miesiączkujących), przynosi również korzyści pacjentkom z zaburzeniami pożądania. Skuteczność takiego postępowania została potwierdzona w badaniach krótko- i długoterminowych, zwłaszcza w przypadku kobiet po menopauzie. Wydaje się, że beneficjentkami takiego postępowania powinny być przede wszystkim pacjentki z niedoczynnością płata przedniego przysadki, jadłowstrętem psychicznym oraz zakażone wirusem HIV. Wszystkie powyższe stany skutkują bowiem równoległym niedoborem androgenów pochodzenia zarówno gonadalnego, jak i nadnerczowego. Te ostatnie ulegają w warunkach fizjologicznych konwersji do silnych androgenów, częściowo poza gonadami, mogąc minimalizować skutki niedostatecznej syntezy testosteronu w gonadach 23.
Najlepiej udokumentowanymi wskazaniami do podawania dehydroepiandrosteronu są zaburzenia pożądania w przebiegu niewydolności nadnerczy (zwłaszcza u kobiet po menopauzie), objawy wynikające z atrofii sromu i pochwy, a zdaniem części autorów również późny hipogonadyzm męski (tzw. adrenopauza), czyli postępujący z wiekiem zanik funkcji hormonalnej warstwy siatkowatej kory nadnerczy.
Tibolon jest lekiem o wielokierunkowym działaniu receptorowym. Spośród trzech głównych jego metabolitów dwa wykazują działanie estrogenowe, trzeci natomiast aktywuje receptor androgenowy i receptor dla progesteronu. Działanie estrogenowe tibolonu jest stwierdzane w mózgu, kościach i nabłonku pochwy, nie obserwuje się go natomiast w endometrium i gruczole sutkowym. Zaletą leku jest zmniejszenie nasilenia objawów menopauzalnych, jeśli towarzyszą one zaburzeniom popędu 24.
Ospemifen jest zaliczany do grupy selektywnych modulatorów receptora estrogenowego. Obok podstawowego wskazania do jego stosowania, jakim jest uczucie suchości w pochwie i dyspareunia, w tych samych dawkach (30–60 mg) jest skuteczny i bezpieczny również w leczeniu zaburzeń pożądania u kobiet w okresie około- i pomenopauzalnym 21. Lek ten wydaje się bezpieczny z punktu widzenia sercowo-naczyniowego, a także sutka i tkanki kostnej.
Oprócz hormonów, w leczeniu zaburzeń pożądania u kobiet stosowane są flibanseryna, sildenafil, arginina, dronabinol, jak również bremelanotyd i bupropion.
Flibanseryna jest agonistą receptora 5-HT1A i równocześnie antagonistą receptora 5-HT2A. W dawce 100 mg poprawia osłabione pożądanie seksualne u kobiet w okresie około- i pomenopauzalnym 21. Jest też skuteczna w leczeniu zaburzeń libido w okresie reprodukcyjnym. Zaleca się przyjmowanie leku w godzinach wieczornych, ponieważ jego stosowanie jest związane ze zwiększonym ryzykiem występowania hipotensji, omdleń oraz depresji OUN 25.
Dane kliniczne dotyczące możliwości poprawy pożądania u kobiet przy użyciu sildenafilu w większych dawkach są niejednoznaczne, jednak w niektórych badaniach wykazano jego korzystny wpływ na obniżone libido u kobiet zarówno z prawidłowym, jak i z obniżonym poziomem estrogenów 21.
W ostatnich latach do leczenia obniżonego libido u kobiet zaproponowano bremelanotyd, będący agonistą receptorów melanokortynowych, z największym powinowactwem do receptorów MC1 i MC4. Pobudzenie drugiego z nich moduluje szlaki nerwowe odpowiedzialne za cykl reakcji seksualnej u kobiet. Stosowanie bremelanotydu powoduje wzrost pożądania, skutkujący wzrostem liczby prób podejmowania aktywności seksualnej, oraz wzrost podniecenia. Lek ten jest rekomendowany dla kobiet z nabytą, uogólnioną postacią zespołu obniżonego popędu seksualnego (HSDD), którą charakteryzuje – obok niskiego pożądania – wyraźny dystres lub zaburzone relacje interpersonalne. Zalecana dawka bremelanotydu wynosi 1,75 mg i powinna być podana podskórnie nie wcześniej niż 45 minut przed planowaną aktywnością seksualną. Lek jest dobrze tolerowany, a do najczęstszych objawów ubocznych należą: nudności (zwłaszcza na początku terapii), uderzenia gorąca, bóle głowy oraz miejscowe reakcje alergiczne. W przeciwieństwie do flibanseryny przy stosowaniu bremelanotydu brakuje przeciwwskazań do stosowania alkoholu w czasie terapii 26. Pewną niedogodność w używaniu bremelanotydu powoduje konieczność jego parenteralnego podawania.
Również bupropion (amfebutamon) w dawce do 300 mg na dobę może poprawiać libido u kobiet w okresie okołomenopauzalnym 27.
Interesującym lekiem do leczenia zaburzeń libido jest syntetyczny kanabinoid – dronabinol. Przyjmowany doraźnie na 1 godzinę przed stosunkiem może zwiększać libido (opis kazuistyczny 28). Lek ten może mieć jednak euforyzujący wpływ na psychikę, a ponadto z uwagi na brak rejestracji w Polsce jego stosowanie ma charakter jedynie eksperymentu medycznego.
Niejasny jest status wyciągu z miłorzębu chińskiego (Ginkgo biloba) w leczeniu zaburzeń pożądania u kobiet. Dotychczasowe badania nie wskazują, żeby był on skuteczny w tym wskazaniu nawet w dawce 300 mg na dobę, ale donoszono również o jego skuteczności. Podobnie istnieją pewne doniesienia dotyczące skuteczności L-argininy w poprawie zaburzeń libido, jednak pochodzą one z badań, w których stosowano preparaty złożone.
W grupie wtórnych – polekowych zaburzeń seksualnych zaburzenia libido mogą być powodowane przez hiperprolaktynemię poneuroleptyczną, kiedy lek przeciwpsychotyczny blokuje receptory dopaminowe w dopaminergicznym układzie podwzgórzowo-lejkowym. W takiej sytuacji skuteczne w leczeniu zaburzeń seksualnych może być zmniejszenie poziomu prolaktyny w surowicy krwi przez podawanie bromokryptyny (tabletki 2,5 mg) w najmniejszej skutecznej dawce. Należy mieć świadomość, że bromokryptyna pobudza receptory dopaminowe typu 2 i działa przeciwstawnie do leku przeciwpsychotycznego. Może więc powodować zarówno osłabienie skuteczności leku przeciwpsychotycznego, jak i efekt psychodysleptyczny. Z uwagi na objawy uboczne bromokryptyny, zwłaszcza zawroty głowy i obniżenie ciśnienia, zasadą jest rozpoczynanie leczenia od małych dawek, podawanych wyjściowo przed snem, a także ich stopniowe zwiększanie, zwykle w odstępach trzydniowych. Z uwagi na krótki okres półtrwania leku jest on podawany 2–3-krotnie w ciągu dnia. W ostatnich latach coraz większe zastsowanie znajduje nowszy i bardziej selektywny agonista receptorów D2 – kabergolina. W porównaniu z bromokryptyną ma dwie istotne zalety: jest lepiej tolerowana, a także – z uwagi na długi okres półtrwania – wymaga podawania raz lub dwa razy w tygodniu. Stosowanie bromokryptyny i kabergoliny z seksuologicznego punktu widzenia wymaga jednak uwzględnienia dwóch niekorzystnych powikłań. Po pierwsze, leki dopaminergiczne mogą powodować tzw. dopa-testotoksykozę, charakteryzującą się hiperseksualnością, której mogą towarzyszyć: uzależnienie od hazardu, kompulsywne robienie zakupów oraz napady przejadania się. Zjawisko to nie zależy od uzyskiwanego w wyniku terapii stężenia prolaktyny i jest tłumaczone nadmierną stymulacją układu mezolimbicznego 29. Drugie z kolei działanie zostało opisane niedawno przez polskich autorów 30, 31 i jest efektem zbyt małego stężenia prolaktyny w czasie terapii bromokryptyną i kabergoliną, związanego ze stosowaniem zbyt dużych dawek tych leków. Polega na spadku pożądania u obu płci, u kobiet dodatkowo zmniejszeniu podniecenia, a u mężczyzn – zaburzeń erekcji. Objawy te mają charakter przejściowy i ustępują w następstwie redukcji dawki agonisty dopaminy powodującej normalizację stężenia prolaktyny. Dlatego w trakcie leczenia uzasadnione wydaje się okresowe monitorowanie stężeń tego hormonu.
Wtórne zaburzenia pożądania seksualnego mogą powodować leki przeciwdepresyjne, szczególnie o działaniu proserotoninowym. Jest to związane ze stymulowaniem przez nie receptorów serotoninowych typu 3 na interneuronach GABA-ergicznych. Stymulacja ta aktywuje interneurony GABA, które z kolei zmniejszają aktywność zarówno neuronów dopaminergicznych w układzie nagrody, jak też noradrenergicznych i serotoninergicznych kontrolujących działanie układu nagrody, z konsekwencją w postaci zmniejszenia uwalniania neuroprzekaźników do szczeliny synaptycznej.
W przypadku leczenia wtórnych zaburzeń libido związanych z działaniem leków proserotoninowych (głównie leki SSRI i wenlafaksyna) leczeniem przyczynowym jest zamiana na lek, który nie wywiera takiego wpływu. Przykładami są wortioksetyna, bupropion, moklobemid, mirtazapina i agomelatyna. W tym aspekcie interesującym lekiem jest wortioksetyna. Sama jest lekiem o mechanizmie działania bliźniaczym do leków z grupy SSRI, jednak, ponieważ blokuje receptory serotoninowe typu 3, nie wywiera depresyjnego wpływu na układ nagrody. Z tego powodu wortioksetyna nie powoduje zaburzeń libido oraz anhedonii.
Bupropion, hamując wychwyt zwrotny noradrenaliny i dopaminy, zwiększa stymulację dopaminergiczną i noradrenergiczną, chroni więc aktywność układu nagrody i zapobiega występowaniu zaburzeń seksualnych. Sam w sobie może być również potencjalnym lekiem do przyczynowego leczenia endogennych zaburzeń libido 32.
Podobnie moklobemid, selektywny i odwracalny inhibitor monoaminooksydazy typu A, może poprawiać libido, zmniejszając rozkład noradrenaliny, a w dużych dawkach wpływając również na zmniejszenie rozkładu dopaminy.
Mirtazapina, podobnie jak mianseryna, blokuje receptory serotoninowe typu 3 oraz 2A, dlatego te leki nawet w małych dawkach, niższych niż minimalna dawka terapeutyczna stosowana do leczenia depresji, mogą być pomocne w korygowaniu zaburzeń seksualnych powodowanych przez lek proserotoninowy.
Wykazano również, że amantadyna w dawce 100–200 mg może być skuteczna w leczeniu zaburzeń pożądania u mężczyzn chorych na schizofrenię, związanych z hiperprolaktynemią, jak również spowodowanych leczeniem przeciwdopaminergicznym bez współistniejącej hiperprolaktynemii 33, 34.
Podsumowanie
Farmakologiczne leczenie zaburzeń seksualnych powinno być zarezerwowane do leczenia pierwotnych zaburzeń seksualnych albo wspomagającego leczenia psychogennych i somatogennych zaburzeń seksualnych. Badania dotyczące farmakologicznego leczenia pożądania opierają się w większości na badaniach kazuistycznych, seriach przypadków i małych grupach pacjentów. Leczenie w każdym przypadku wymaga personalizacji, a w sytuacji braku poprawy podejmowania kolejnych prób z wykorzystaniem danych pochodzących z publikacji klinicznych.
Piśmiennictwo:
1. Giles KR, McCabe MP. Conceptualizing women’s sexual function: linear vs. circular models of sexual response. J Sex Med 2009; 6: 2761-2771.
2. Giraldi A, Kristensen E, Sand M. Endorsement of models describing sexual response of men and women with a sexual partner: an online survey in a population sample of Danish adults ages 20-65 years. J Sex Med 2015; 12: 116-128.
3. Carvalho J, Vieira A, Nobre P. Latent structures of male sexual functioning. J Sex Med 2011; 8: 2501-2511.
4. Hoshina Y, Takeo T, Nakano K i wsp. Axon-sparing lesion of the preoptic area enhances receptivity and diminishes proceptivity among components of female rat sexual behavior. Behav Brain Res 1994: 61: 247-279.
5. Hull EM, Lorrain DS, Du J i wsp. Hormone neurotransmitter interactions in the control of sexual behavior. Behav Brain Res 1999; 105: 105-115.
6. Meston CM, Gorzalka BB, Wright JM. Inhibition of physiological and subjective sexual arousal in women by clonidine. Psychosom Med 1997; 59: 399-407.
7. Caldwell JD, Jirikowski GF, Greer ER i wsp. Medial preoptic area and female sexual receptivity. Behav Neurosci 1989; 103: 655-662.
8. Succu S, Sanna F, Melis T i wsp. Stimulation of dopamine receptors in the paraventricular nucleus of the hypothalamus of the male rats induces penile erection and increases extra-cellular dopamine in the nucleus accumbens. Neuropharmacology 2007; 52: 1034-1043.
9. Oosterom J, Nijenhuis WA, Schaaper WM i wsp. Conformation of the core sequence in melanocortin peptides directs selectivity for the melanocortin MC3 and MC4 receptors. J Biol Chem 1999; 274: 16853-16860.
10. Medina F, Siddiqui A, Scimonelli T i wsp. The inter-relationship between gonadal steroids and POMC peptides, beta-endorphin and alpha-MSH in the control of sexual behavior in the female rat. Peptides 1998; 19: 1309-1316.
11. Just MJ. The influence of atypical antipsychotic drugs on sexual function. Neuropsychiatr Dis Treat 2015; 11: 1655-1661.
12. Marson L, McKenna KE. Serotonergic neurotoxic lesions facilitate male sexual reflexes. Pharmacol Biochem Behav 1994; 47: 883-888.
13. Klint T, Dahlgren IL, Larsson K. The selective 5-HT2 receptor antagonist amperozide attenuates 1-(2,5-dimethoxy-4-iodophenyl)-2--aminopropane-induced inhibition of male rat sexual behavior. Eur J Pharmacol 1992; 212: 241-246.
14. Band LC, Hull EM. Morphine and dynorphin (1-13) microinjected into the medial preoptic area and nucleus accumbens: effects on sexual behavior in male rats. Brain Res 1990; 524: 77-84.
15. Rodriguez-Manzo G, Asai M, Fernandez-Guasti A. Evidence for changes in brain enkephalin contents associated to male rat sexual activity. Behav Brain Res 2002; 131: 47-55.
16. Mitchell JB, Stewart J. Facilitation of sexual behaviors in the male rat associated with intra-VTA injections of opiates. Pharmacol Biochem Behav 1990; 35: 643-650.
17. Acosta-Martinez M, Etgen AM. The role of delta-opioid receptors in estrogen facilitation of lordosis behavior. Behav Brain Res 2002; 136: 93-102.
18. Bradshaw HB, Rimmerman N, Krey JF i wsp. Sex and hormonal cycle differences in rat brain levels of pain-related cannabimimetic lipid mediators. Am J Physiol Regul Integr Comp Physiol 2006; 291: 349-358.
19. Ye Y, Lu Z, Zhou W. Pheromone effects on the human hypothalamus in relation to sexual orientation and gender. Handb Clin Neurol 2021; 182: 293-306.
20. Micevych PE, Meisel RL. Integrating neural circuits controlling female sexual behavior. Front Syst Neurosci 2017; 11: 42.
21. Clayton AH, Valladares-Juarez EM. Female sexual dysfunction. Med Clin North Am 2019; 103: 681-698.
22. Rastrelli G, Corona G, Maggi M. Testosterone and sexual function in men. Maturitas 2018; 112: 46-52.
23. Krysiak R, Okopień B. Niedobór androgenów u kobiet. Wiad Lek 2013; 66: 360-369.
24. Del Río JP, Molina S, Hidalgo-Lanussa O i wsp. Tibolone as hormonal therapy and neuroprotective agent. Trends Endocrinol Metab 2020; 31: 742-759.
25. Clements JN, Thompson B. Flibanserin for hypoactive sexual desire disorder in premenopausal women. JAAPA 2018; 31: 51-53.
26. Dhillon S, Keam SJ. Bremelanotide: first approval. Drugs 2019; 79: 1599-1606.
27. Segraves RT, Clayton A, Croft H i wsp. Bupropion sustained release for the treatment of hypoactive sexual desire disorder in premenopausal women. J Clin Psychopharmacol 2004; 24: 339-342.
28. Salerian AJ. Successful treatment of sexual dysfunction with dronabinol: a case report. J Clin Psychiatry 2004; 65: 1146-1147.
29. De Sousa SM, Chapman IM, Falhammar H i wsp. Dopa-testotoxicosis: disruptive hypersexuality in hypogonadal men with prolactinomas treated with dopamine agonists. Endocrine 2017; 55: 618-624.
30. Krysiak R, Kowalcze K, Okopień B. Sexual function and depressive symptoms in young women with hypoprolactinaemia. Clin Endocrinol (Oxf) 2020; 93: 482-488.
31. Krysiak R, Kowalcze K, Okopień B. Sexual function and depressive symptoms in men with hypoprolactinaemia secondary to overtreatment of prolactin excess: a pilot study. Endocrinol Diabetes Nutr (Engl Ed) 2021; S2530-0164(21): 00145-2.
32. Simonsen U, Comerma-Steffensen S, Andersson KE. Modulation of dopaminergic pathways to treat erectile dysfunction. Basic Clin Pharmacol Toxicol 2016; 119 (Suppl 3): 63-74.
33. Valevski A, Modai I, Zbarski E i wsp. Effect of amantadine on sexual dysfunction in neuroleptic-treated male schizophrenic patients. Clin Neuropharmacol 1998; 21: 355-357.
34. Krzystanek M, Warchala A, Trędzbor B i wsp. Amantadine in the treatment of sexual inactivity in schizophrenia patients taking atypical antipsychotics – the pilot case series study. Pharmaceuticals (Basel) 2021; 14: 947.
Tekst pochodzi z „Psychiatrii Spersonalizowanej” 1/2022. Czasopismo można zamówić na stronie: www.termedia.pl/ps/prenumerata.

Podstawą prawidłowych funkcji seksualnych jest brak patologii w budowie i funkcjonowaniu części układu nerwowego związanych z reakcją seksualną i w narządach płciowych, prawidłowa czynność gonad z prawidłowym stężeniem estrogenów, progesteronu i testosteronu oraz brak zaburzeń somatycznych, psychologicznych i psychiatrycznych, mogących zakłócać funkcje seksualne, albo jak w przypadku problemów medycznych – konieczność stosowania leków z objawami niepożądanymi w postaci zaburzeń seksualnych. W leczeniu zaburzeń seksualnych możliwe są różne strategie, obejmujące psychoterapię, farmakoterapię/hormonoterapię, modyfikację leczenia farmakologicznego powodującego objawy niepożądane w postaci dysfunkcji seksualnych lub metody zabiegowe. Autorzy dokonali przeglądu badań klinicznych, poszukując danych dotyczących farmakologicznego leczenia zaburzeń pożądania. W aspekcie farmakologicznego leczenia zaburzeń pożądania większość badań ma charakter opisów przypadków, serii przypadków i badań przeprowadzonych na małych grupach osób. Farmakologiczne leczenie zaburzeń pożądania powinno być zarezerwowane do leczenia pierwotnych zaburzeń seksualnych albo wspomagającego leczenia psychogennych i somatogennych zaburzeń. Leczenie w każdym przypadku wymaga personalizacji, a w sytuacji braku poprawy podejmowania kolejnych prób z wykorzystaniem dostępnych danych z publikacji klinicznych.
Wstęp
Współczesnej seksuologii udało się opisać fizjologiczny, liniowy wzorzec reakcji seksualnej człowieka, będący podstawą do opisu nieprawidłowości pojawiających się w przebiegu stosunku. Według tego modelu reakcja seksualna przebiega u kobiet i mężczyzn bez zaburzeń seksualnych przez kolejne fazy pożądania, podniecenia, plateau, orgazmu i odprężenia. Linearny model reakcji seksualnej nie został dotąd sfalsyfikowany i jest nadal podstawą do opisywania zaburzeń seksualnych człowieka, jako odchyleń od tej fizjologii 1, 2.
W linearnym modelu zaburzeń seksualnych kluczowe wydaje się pożądanie seksualne. W odniesieniu do pożądania seksualnego, obecnie rozróżnia się dwa jego rodzaje: spontaniczne i reaktywne, przy czym pożądanie u mężczyzn może nie występować albo jest tożsame z uświadamianym podnieceniem seksualnym 3. Brak pożądania (lub pożądania utożsamianego z podnieceniem) uniemożliwia fizyczną reakcję podniecenia i odbycie stosunku seksualnego.
Przyczyny zaburzeń pożądania są złożone i mogą mieć charakter psychologiczny i/lub somatyczny. W leczeniu tych zaburzeń możliwe są różne strategie, obejmujące psychoterapię, farmakoterapię/hormonoterapię, modyfikację leczenia powodującego objawy niepożądane w postaci dysfunkcji seksualnych lub metody zabiegowe.
Celem niniejszego artykułu jest z jednej strony przedstawienie biologicznych uwarunkowań pożądania, a z drugiej – omówienie dostępnych doniesień na temat możliwości farmakologicznej korekcji zaburzeń libido. Informacje te mogą mieć praktyczne znaczenie dla lekarzy szukających farmakologicznych sposobów poprawy libido u kobiet i mężczyzn.
Biologiczne uwarunkowania libido
Libido jest złożoną funkcją seksualną, na którą wpływają czynniki psychologiczne i biologiczne. Neurofizjologiczne tło zaburzeń pożądania stanowią przede wszystkim dysfunkcje układu nagrody, a w szczególności budujących go struktur układu mezolimbicznego. Wchodzące w jego skład szlaki regulatorowe kontrolowane są przez neurony układu serotoninergicznego i noradrenergicznego, syntezowane miejscowo i obwodowo hormony neuropeptydowe, u kobiet przez estrogeny i progesteron, u mężczyzn natomiast przez poziom testosteronu. Liczne badania podstawowe oparte na modelach zwierzęcych oraz obserwacje kliniczne umożliwiły identyfikację endogennych czynników pobudzających i hamujących reakcje seksualne (rycina 1).
Inicjujące stan pożądania neurony dopaminergiczne systemu mezolimbicznego zlokalizowane są w polu brzusznym nakrywki (VTA), skąd oddają swe projekcje do szeregu struktur podkorowych, w szczególności do ciała migdałowatego i jądra półleżącego. Istotną rolę w generowaniu reakcji seksualnych przypisuje się również neuronom dopaminergicznym warstwy niepewnej (zona incerta), których aksony docierają do przyśrodkowego pola przedwzrokowego podwzgórza (mPOA). Efektem selektywnego zniszczenia zarówno mPOA, jak i jądra półleżącego w mózgu szczura jest daleko idąca redukcja zachowań seksualnych 4. Rola dopaminy w inicjowaniu pobudzenia seksualnego leży u podstaw wpływu leków neuropsychiatrycznych na stan pożądania u ludzi. Przykładowo L-DOPA (3,4-dihydroksy-L-fenyloalanina) wykazuje działanie stymulujące reakcje seksualne, które może być wygaszane działaniem nieselektywnych antagonistów receptorów dopaminergicznych. W związku z tym współcześnie stosowane leki antypsychotyczne, będące w większości antagonistami transmisji dopaminowej, mogą zaburzać szeroko rozumianą aktywność seksualną obserwowaną zarówno w modelach zwierzęcych, jak i w badaniach klinicznych. Niewykluczone, że kluczową rolę w dopaminergicznej stymulacji seksualnej u zwierząt może odgrywać ilościowy stosunek receptorów D1 względem D2 w obszarze mPOA. Zgodnie z tym modelem, aktywacja receptora D1 wyzwala psychiczne składowe pożądania, natomiast stymulacja receptora D2 związana jest raczej czynnością wykonawczą układu płciowego 5. Szlaki noradrenergiczne mózgowia, których głównym źródłem są neurony miejsca sinawego (locus coeruleus) pnia mózgu, inicjują pobudzenie seksualne i podtrzymują je drogą stymulacji układu współczulnego. Oddają one liczne projekcje zaopatrujące podwzgórze, układ limbiczny i pewne okolice kory nowej. Agoniści receptora α2, jak klonidyna, zmniejszają uwalnianie neurotransmitera, czego efektem jest depresja układu współczulnego i obniżenie wrażliwości na bodźce o charakterze erotycznym 6, antagoniści natomiast, m.in. johimbina, wywołują efekt przeciwny, dlatego bywają niekiedy stosowane jako stymulanty seksualne. Inhibitory syntezy noradrenaliny, np. dietyloditiokarbaminian, wyciszają przejawy podniecenia seksualnego u zwierząt. Neurohormonem włączonym w ośrodkowe mechanizmy generowania podniecenia seksualnego jest również oksytocyna, nonapeptyd syntezowany w wielkokomórkowych jądrach neurosekrecyjnych podwzgórza. Iniekcja oksytocyny do mPOA lub jądra brzuszno-przyśrodkowego podwzgórza (VMH) szczurów powoduje w przypadku samic wzmożenie behawioralnych efektów pobudzenia seksualnego (lordoza), natomiast u samców stymuluje erekcję 7. Stymulacja receptorów dopaminergicznych D1 w obrębie jądra przykomorowego podwzgórza (PVN) powoduje zarówno uwolnienie oksytocyny, jak i wzrost poziomu dopaminy w jądrze półleżącym, co sugeruje obecność zależnego od oksytocyny systemu integrującego podwzgórzowy i mezolimbiczny szlak dopaminergiczny 8. Kolejnymi neuropeptydami wyzwalającymi stan pożądania i pobudzenia seksualnego są dwie pochodne proopiomelanokortyny (POMC): adrenokortykotropina (ACTH) oraz melanotropina (α-MSH), których cząsteczki wiążą się ze swoistymi receptorami melanokortynowymi MC3 i MC4 licznych neuronów podwzgórza i układu limbicznego 9. Źródłem melanokortyn są neurony jądra łukowatego podwzgórza, oddające liczne rozproszone projekcje zarówno do struktur sąsiadujących, jak i odległych, w tym układu limbicznego, śródmózgowia i pnia mózgu. Neurony te wykazują wrażliwość na steroidy, zaobserwowano bowiem podwyższenie poziomu α-MSH w podwzgórzu samic szczurzych po podaniu estradiolu 10. Farmakomodulacja receptorów melanokortynowych (MCRs) wydaje się niezwykle obiecującą strategią terapeutyczną w zaburzeniach seksualnych.
Kluczową rolę w hamowaniu stanu pożądania odgrywają szlaki serotoninergiczne biorące swój początek w śródmózgowiowych jądrach szwu (nuclei raphes). Aksony opuszczające tę strukturę docierają do licznych okolic ośrodkowego układu nerwowego (OUN): kresomózgowia, podwzgórza, układu limbicznego, hipokampa, pnia mózgu oraz eferentnie do rdzenia kręgowego, gdzie zaopatrują autonomiczne ośrodki w lędźwiowej i krzyżowej części rdzenia kręgowego, kontrolujące odruchy genitalne. Co warte podkreślenia, serotonina hamuje funkcje seksualne drogą stymulacji receptorów 5-HT2A i 5-HT2C, efektem aktywacji autoreceptorów 5-HT1A jest natomiast wzrost pożądania i redukcja uwalniania neurotransmitera do szczeliny synaptycznej 11. Neurotoksyczne uszkodzenie zstępujących dróg serotoninergicznych rdzenia kręgowego silnie pobudza erekcję, co sugeruje, że odruch wzwodu jest stale tonicznie hamowany przez te szlaki 12. Istotne opóźnienie ejakulacji oraz anorgazmia to efekty bardzo często obserwowane u pacjentów przyjmujących leki przeciwdepresyjne z grupy selektywnych inhibitorów wychwytu zwrotnego serotoniny (SSRIs). Zjawiska te mogą być, przynajmniej częściowo, odwrócone przez podanie oksytocyny. Szczury poddane działaniu DOI (2,5-dimetoksy-4-jodoamfetamina), antagonisty receptorów 5-HT1C i 5-HT2, manifestowały zahamowaną aktywność seksualną 13. Zastosowanie psychoaktywnego agonisty receptorów 5-HT1C i 5-HT2 – TFMPP (3-trifluorometylofenylopiperazyna) – powodowało zniesienie odruchów kopulacyjnych u samców królików. Ważnymi inhibitorami pobudzenia seksualnego są endogenne peptydy opioidowe, zarówno pochodne POMC, takie jak β-endorfna (β-END), jak i proenkefaliny (Met- i Leu- enkefalina) oraz prodynorfiny (dynorfiny A i B), manifestujące zróżnicowane powinowactwo do receptorów opioidowych μ, δ i κ. Głównym źródłem opioidów są neurony POMC jądra łukowatego podwzgórza wysyłające liczne projekcje do kresomózgowia, śródmózgowia, pola brzusznego nakrywki, jąder półleżących i prążkowia. Podanie agonistów receptora opioidowego μ do przyśrodkowego mPOA lub VMH podwzgórza hamuje zachowania seksualne szczurów 14. Prawdopodobnie uwolnienie opioidów w mPOA aktywuje układ nagrody i jest czynnikiem generującym stan refrakcji seksualnej 15. Z odmienną sytuacją mamy do czynienia w przypadku VTA, gdzie uwolnienie opioidów pociąga za sobą stymulację aktywności seksualnej, a celowana infuzja morfiny lub dynorfiny do tej okolicy wyzwala szereg behawioralnych symptomów pobudzenia seksualnego u szczurów obydwu płci 16. Dokomorowa iniekcja agonistów receptora δ skutkuje uaktywnieniem szeregu zachowań seksualnych, natomiast iniekcja do mPOA wywołuje efekt przeciwny 17. Substancjami wygaszającymi aktywność seksualną są również endogenne kanabinoidy, m.in. anandamid (arachidonyloetanoloamina, AEA) czy 2-arachidonyloglicerol (2-AG), działające za pośrednictwem receptora kanabinoidowego typu 1 (CB1) obecnego w licznych strukturach podwzgórza i układu limbicznego. Najwyższe stężenie endogennych kanabinoidów w podwzgórzu stwierdzono u samic szczura w okresie międzyrujowym (diestrus), kiedy aktywność seksualna zwierząt jest istotnie obniżona, po czym w miarę przechodzenia w kolejne fazy cyklu rozrodczego następował jego stopniowy spadek 18. Hormonem hamującym reakcje seksualne, w tym stany pożądania i ekscytacji, jest również prolaktyna, peptyd produkowany przez komórki kwasochłonne przysadki gruczołowej, odgrywający wiodącą rolę w regulacji cyklu jajnikowego i procesu laktacji. Podstawowym inhibitorem sekrecji prolaktyny jest dopamina. Blokada transmisji dopaminergicznej prowadzi zatem do hiperprolaktynemii, której efektem jest stymulacja hamującej sygnalizacji GABA-ergicznej i opioidowej, co istotnie obniża poziom libido. Najnowsze badania sugerują również wpływ endogennych steroidów o potencjalnych cechach ludzkich feromonów płciowych: androsta-4,16,-dien-3-onu oraz estra-1,3,5(10),16-tetraen-3-olu na podwzgórzowe mechanizmy pożądania i podniecenia seksualnego. Ich efekty fizjologiczne wydają się zależne od płci i orientacji seksualnej 19. Podwzgórzowo-limbiczne mechanizmy pożądania podlegają pewnym odmiennościom ze względu na płeć. Na rycinie 2 przedstawiono przykładowo regulację pożądania u kobiet.
Farmakoterapia zaburzeń libido
Zaburzenia libido mogą mieć charakter pierwotny albo wtórny, spowodowany np. zażywanymi lekami albo zaburzeniami hormonalnymi. W sytuacji potwierdzonych zaburzeń poziomu hormonów płciowych we krwi, w leczeniu zaburzeń libido stosuje się miejscową lub systemową substytucję hormonalną estrogenami, substytucję systemową testosteronem lub progesteronami albo podawanie selektywnego modulatora receptora estrogenowego – tibolonu lub ospemifenu.
Hormonalna terapią zastępcza (HTZ) ma mały do średniego wpływ na poprawę pożądania i podniecenia u kobiet w okresie około- i pomenopauzalnym. Odgrywa natomiast ważną rolą w przywróceniu troficzności nabłonka pochwy, likwidując nieprzyjemne odczucia przy stosunku czy dyspareunię 21.
W leczeniu zaburzeń libido u kobiet poprawę może przynieść podawanie androgenów, zwłaszcza w przypadku małego stężenia całkowitego testosteronu i siarczanu dehydroepiandrosteronu. Jeszcze szerzej wykorzystuje się leczenie za pomocą androgenów zaburzeń pożądania u mężczyzn, pod warunkiem że ich stężenie mieści poniżej normy dla wieku. W większości krajów europejskich i w USA dostępne są różne formy podawania testosteronu (tabletki, iniekcje domięśniowe, kremy, żele, plastry, tabletki dopoliczkowe, aerozol do aplikacji pod pachę), przy czym stosowane dawki są znacznie większe niż dla kobiet, co odzwierciedla fizjologiczne różnice w dobowym wytwarzaniu tego hormonu (około 5–10 mg u mężczyzn i 150–300 μg u kobiet) 21, 22. Natomiast egzogenny dehydroepiandrosteron (prasteron) jest stosowany w zbliżonych dawkach u obu płci, przy czym w leczeniu zmniejszonego libido preferuje się doustną drogę podaży leku.
Substytucja testosteronu u mężczyzn może poprawiać zaburzenia pożądania, jednak – jak wspomniano – jest ona skuteczna praktycznie wyłącznie w przypadkach potwierdzonego zmniejszenia stężenia tego hormonu w osoczu 22. Podawanie testosteronu kobietom, zwykle w postaci plastrów uwalniających 300 μg hormonu na dobę (dawka umożliwiająca uzyskanie stężenia testosteronu w górnych granicach normy dla kobiet miesiączkujących), przynosi również korzyści pacjentkom z zaburzeniami pożądania. Skuteczność takiego postępowania została potwierdzona w badaniach krótko- i długoterminowych, zwłaszcza w przypadku kobiet po menopauzie. Wydaje się, że beneficjentkami takiego postępowania powinny być przede wszystkim pacjentki z niedoczynnością płata przedniego przysadki, jadłowstrętem psychicznym oraz zakażone wirusem HIV. Wszystkie powyższe stany skutkują bowiem równoległym niedoborem androgenów pochodzenia zarówno gonadalnego, jak i nadnerczowego. Te ostatnie ulegają w warunkach fizjologicznych konwersji do silnych androgenów, częściowo poza gonadami, mogąc minimalizować skutki niedostatecznej syntezy testosteronu w gonadach 23.
Najlepiej udokumentowanymi wskazaniami do podawania dehydroepiandrosteronu są zaburzenia pożądania w przebiegu niewydolności nadnerczy (zwłaszcza u kobiet po menopauzie), objawy wynikające z atrofii sromu i pochwy, a zdaniem części autorów również późny hipogonadyzm męski (tzw. adrenopauza), czyli postępujący z wiekiem zanik funkcji hormonalnej warstwy siatkowatej kory nadnerczy.
Tibolon jest lekiem o wielokierunkowym działaniu receptorowym. Spośród trzech głównych jego metabolitów dwa wykazują działanie estrogenowe, trzeci natomiast aktywuje receptor androgenowy i receptor dla progesteronu. Działanie estrogenowe tibolonu jest stwierdzane w mózgu, kościach i nabłonku pochwy, nie obserwuje się go natomiast w endometrium i gruczole sutkowym. Zaletą leku jest zmniejszenie nasilenia objawów menopauzalnych, jeśli towarzyszą one zaburzeniom popędu 24.
Ospemifen jest zaliczany do grupy selektywnych modulatorów receptora estrogenowego. Obok podstawowego wskazania do jego stosowania, jakim jest uczucie suchości w pochwie i dyspareunia, w tych samych dawkach (30–60 mg) jest skuteczny i bezpieczny również w leczeniu zaburzeń pożądania u kobiet w okresie około- i pomenopauzalnym 21. Lek ten wydaje się bezpieczny z punktu widzenia sercowo-naczyniowego, a także sutka i tkanki kostnej.
Oprócz hormonów, w leczeniu zaburzeń pożądania u kobiet stosowane są flibanseryna, sildenafil, arginina, dronabinol, jak również bremelanotyd i bupropion.
Flibanseryna jest agonistą receptora 5-HT1A i równocześnie antagonistą receptora 5-HT2A. W dawce 100 mg poprawia osłabione pożądanie seksualne u kobiet w okresie około- i pomenopauzalnym 21. Jest też skuteczna w leczeniu zaburzeń libido w okresie reprodukcyjnym. Zaleca się przyjmowanie leku w godzinach wieczornych, ponieważ jego stosowanie jest związane ze zwiększonym ryzykiem występowania hipotensji, omdleń oraz depresji OUN 25.
Dane kliniczne dotyczące możliwości poprawy pożądania u kobiet przy użyciu sildenafilu w większych dawkach są niejednoznaczne, jednak w niektórych badaniach wykazano jego korzystny wpływ na obniżone libido u kobiet zarówno z prawidłowym, jak i z obniżonym poziomem estrogenów 21.
W ostatnich latach do leczenia obniżonego libido u kobiet zaproponowano bremelanotyd, będący agonistą receptorów melanokortynowych, z największym powinowactwem do receptorów MC1 i MC4. Pobudzenie drugiego z nich moduluje szlaki nerwowe odpowiedzialne za cykl reakcji seksualnej u kobiet. Stosowanie bremelanotydu powoduje wzrost pożądania, skutkujący wzrostem liczby prób podejmowania aktywności seksualnej, oraz wzrost podniecenia. Lek ten jest rekomendowany dla kobiet z nabytą, uogólnioną postacią zespołu obniżonego popędu seksualnego (HSDD), którą charakteryzuje – obok niskiego pożądania – wyraźny dystres lub zaburzone relacje interpersonalne. Zalecana dawka bremelanotydu wynosi 1,75 mg i powinna być podana podskórnie nie wcześniej niż 45 minut przed planowaną aktywnością seksualną. Lek jest dobrze tolerowany, a do najczęstszych objawów ubocznych należą: nudności (zwłaszcza na początku terapii), uderzenia gorąca, bóle głowy oraz miejscowe reakcje alergiczne. W przeciwieństwie do flibanseryny przy stosowaniu bremelanotydu brakuje przeciwwskazań do stosowania alkoholu w czasie terapii 26. Pewną niedogodność w używaniu bremelanotydu powoduje konieczność jego parenteralnego podawania.
Również bupropion (amfebutamon) w dawce do 300 mg na dobę może poprawiać libido u kobiet w okresie okołomenopauzalnym 27.
Interesującym lekiem do leczenia zaburzeń libido jest syntetyczny kanabinoid – dronabinol. Przyjmowany doraźnie na 1 godzinę przed stosunkiem może zwiększać libido (opis kazuistyczny 28). Lek ten może mieć jednak euforyzujący wpływ na psychikę, a ponadto z uwagi na brak rejestracji w Polsce jego stosowanie ma charakter jedynie eksperymentu medycznego.
Niejasny jest status wyciągu z miłorzębu chińskiego (Ginkgo biloba) w leczeniu zaburzeń pożądania u kobiet. Dotychczasowe badania nie wskazują, żeby był on skuteczny w tym wskazaniu nawet w dawce 300 mg na dobę, ale donoszono również o jego skuteczności. Podobnie istnieją pewne doniesienia dotyczące skuteczności L-argininy w poprawie zaburzeń libido, jednak pochodzą one z badań, w których stosowano preparaty złożone.
W grupie wtórnych – polekowych zaburzeń seksualnych zaburzenia libido mogą być powodowane przez hiperprolaktynemię poneuroleptyczną, kiedy lek przeciwpsychotyczny blokuje receptory dopaminowe w dopaminergicznym układzie podwzgórzowo-lejkowym. W takiej sytuacji skuteczne w leczeniu zaburzeń seksualnych może być zmniejszenie poziomu prolaktyny w surowicy krwi przez podawanie bromokryptyny (tabletki 2,5 mg) w najmniejszej skutecznej dawce. Należy mieć świadomość, że bromokryptyna pobudza receptory dopaminowe typu 2 i działa przeciwstawnie do leku przeciwpsychotycznego. Może więc powodować zarówno osłabienie skuteczności leku przeciwpsychotycznego, jak i efekt psychodysleptyczny. Z uwagi na objawy uboczne bromokryptyny, zwłaszcza zawroty głowy i obniżenie ciśnienia, zasadą jest rozpoczynanie leczenia od małych dawek, podawanych wyjściowo przed snem, a także ich stopniowe zwiększanie, zwykle w odstępach trzydniowych. Z uwagi na krótki okres półtrwania leku jest on podawany 2–3-krotnie w ciągu dnia. W ostatnich latach coraz większe zastsowanie znajduje nowszy i bardziej selektywny agonista receptorów D2 – kabergolina. W porównaniu z bromokryptyną ma dwie istotne zalety: jest lepiej tolerowana, a także – z uwagi na długi okres półtrwania – wymaga podawania raz lub dwa razy w tygodniu. Stosowanie bromokryptyny i kabergoliny z seksuologicznego punktu widzenia wymaga jednak uwzględnienia dwóch niekorzystnych powikłań. Po pierwsze, leki dopaminergiczne mogą powodować tzw. dopa-testotoksykozę, charakteryzującą się hiperseksualnością, której mogą towarzyszyć: uzależnienie od hazardu, kompulsywne robienie zakupów oraz napady przejadania się. Zjawisko to nie zależy od uzyskiwanego w wyniku terapii stężenia prolaktyny i jest tłumaczone nadmierną stymulacją układu mezolimbicznego 29. Drugie z kolei działanie zostało opisane niedawno przez polskich autorów 30, 31 i jest efektem zbyt małego stężenia prolaktyny w czasie terapii bromokryptyną i kabergoliną, związanego ze stosowaniem zbyt dużych dawek tych leków. Polega na spadku pożądania u obu płci, u kobiet dodatkowo zmniejszeniu podniecenia, a u mężczyzn – zaburzeń erekcji. Objawy te mają charakter przejściowy i ustępują w następstwie redukcji dawki agonisty dopaminy powodującej normalizację stężenia prolaktyny. Dlatego w trakcie leczenia uzasadnione wydaje się okresowe monitorowanie stężeń tego hormonu.
Wtórne zaburzenia pożądania seksualnego mogą powodować leki przeciwdepresyjne, szczególnie o działaniu proserotoninowym. Jest to związane ze stymulowaniem przez nie receptorów serotoninowych typu 3 na interneuronach GABA-ergicznych. Stymulacja ta aktywuje interneurony GABA, które z kolei zmniejszają aktywność zarówno neuronów dopaminergicznych w układzie nagrody, jak też noradrenergicznych i serotoninergicznych kontrolujących działanie układu nagrody, z konsekwencją w postaci zmniejszenia uwalniania neuroprzekaźników do szczeliny synaptycznej.
W przypadku leczenia wtórnych zaburzeń libido związanych z działaniem leków proserotoninowych (głównie leki SSRI i wenlafaksyna) leczeniem przyczynowym jest zamiana na lek, który nie wywiera takiego wpływu. Przykładami są wortioksetyna, bupropion, moklobemid, mirtazapina i agomelatyna. W tym aspekcie interesującym lekiem jest wortioksetyna. Sama jest lekiem o mechanizmie działania bliźniaczym do leków z grupy SSRI, jednak, ponieważ blokuje receptory serotoninowe typu 3, nie wywiera depresyjnego wpływu na układ nagrody. Z tego powodu wortioksetyna nie powoduje zaburzeń libido oraz anhedonii.
Bupropion, hamując wychwyt zwrotny noradrenaliny i dopaminy, zwiększa stymulację dopaminergiczną i noradrenergiczną, chroni więc aktywność układu nagrody i zapobiega występowaniu zaburzeń seksualnych. Sam w sobie może być również potencjalnym lekiem do przyczynowego leczenia endogennych zaburzeń libido 32.
Podobnie moklobemid, selektywny i odwracalny inhibitor monoaminooksydazy typu A, może poprawiać libido, zmniejszając rozkład noradrenaliny, a w dużych dawkach wpływając również na zmniejszenie rozkładu dopaminy.
Mirtazapina, podobnie jak mianseryna, blokuje receptory serotoninowe typu 3 oraz 2A, dlatego te leki nawet w małych dawkach, niższych niż minimalna dawka terapeutyczna stosowana do leczenia depresji, mogą być pomocne w korygowaniu zaburzeń seksualnych powodowanych przez lek proserotoninowy.
Wykazano również, że amantadyna w dawce 100–200 mg może być skuteczna w leczeniu zaburzeń pożądania u mężczyzn chorych na schizofrenię, związanych z hiperprolaktynemią, jak również spowodowanych leczeniem przeciwdopaminergicznym bez współistniejącej hiperprolaktynemii 33, 34.
Podsumowanie
Farmakologiczne leczenie zaburzeń seksualnych powinno być zarezerwowane do leczenia pierwotnych zaburzeń seksualnych albo wspomagającego leczenia psychogennych i somatogennych zaburzeń seksualnych. Badania dotyczące farmakologicznego leczenia pożądania opierają się w większości na badaniach kazuistycznych, seriach przypadków i małych grupach pacjentów. Leczenie w każdym przypadku wymaga personalizacji, a w sytuacji braku poprawy podejmowania kolejnych prób z wykorzystaniem danych pochodzących z publikacji klinicznych.
Piśmiennictwo:
1. Giles KR, McCabe MP. Conceptualizing women’s sexual function: linear vs. circular models of sexual response. J Sex Med 2009; 6: 2761-2771.
2. Giraldi A, Kristensen E, Sand M. Endorsement of models describing sexual response of men and women with a sexual partner: an online survey in a population sample of Danish adults ages 20-65 years. J Sex Med 2015; 12: 116-128.
3. Carvalho J, Vieira A, Nobre P. Latent structures of male sexual functioning. J Sex Med 2011; 8: 2501-2511.
4. Hoshina Y, Takeo T, Nakano K i wsp. Axon-sparing lesion of the preoptic area enhances receptivity and diminishes proceptivity among components of female rat sexual behavior. Behav Brain Res 1994: 61: 247-279.
5. Hull EM, Lorrain DS, Du J i wsp. Hormone neurotransmitter interactions in the control of sexual behavior. Behav Brain Res 1999; 105: 105-115.
6. Meston CM, Gorzalka BB, Wright JM. Inhibition of physiological and subjective sexual arousal in women by clonidine. Psychosom Med 1997; 59: 399-407.
7. Caldwell JD, Jirikowski GF, Greer ER i wsp. Medial preoptic area and female sexual receptivity. Behav Neurosci 1989; 103: 655-662.
8. Succu S, Sanna F, Melis T i wsp. Stimulation of dopamine receptors in the paraventricular nucleus of the hypothalamus of the male rats induces penile erection and increases extra-cellular dopamine in the nucleus accumbens. Neuropharmacology 2007; 52: 1034-1043.
9. Oosterom J, Nijenhuis WA, Schaaper WM i wsp. Conformation of the core sequence in melanocortin peptides directs selectivity for the melanocortin MC3 and MC4 receptors. J Biol Chem 1999; 274: 16853-16860.
10. Medina F, Siddiqui A, Scimonelli T i wsp. The inter-relationship between gonadal steroids and POMC peptides, beta-endorphin and alpha-MSH in the control of sexual behavior in the female rat. Peptides 1998; 19: 1309-1316.
11. Just MJ. The influence of atypical antipsychotic drugs on sexual function. Neuropsychiatr Dis Treat 2015; 11: 1655-1661.
12. Marson L, McKenna KE. Serotonergic neurotoxic lesions facilitate male sexual reflexes. Pharmacol Biochem Behav 1994; 47: 883-888.
13. Klint T, Dahlgren IL, Larsson K. The selective 5-HT2 receptor antagonist amperozide attenuates 1-(2,5-dimethoxy-4-iodophenyl)-2--aminopropane-induced inhibition of male rat sexual behavior. Eur J Pharmacol 1992; 212: 241-246.
14. Band LC, Hull EM. Morphine and dynorphin (1-13) microinjected into the medial preoptic area and nucleus accumbens: effects on sexual behavior in male rats. Brain Res 1990; 524: 77-84.
15. Rodriguez-Manzo G, Asai M, Fernandez-Guasti A. Evidence for changes in brain enkephalin contents associated to male rat sexual activity. Behav Brain Res 2002; 131: 47-55.
16. Mitchell JB, Stewart J. Facilitation of sexual behaviors in the male rat associated with intra-VTA injections of opiates. Pharmacol Biochem Behav 1990; 35: 643-650.
17. Acosta-Martinez M, Etgen AM. The role of delta-opioid receptors in estrogen facilitation of lordosis behavior. Behav Brain Res 2002; 136: 93-102.
18. Bradshaw HB, Rimmerman N, Krey JF i wsp. Sex and hormonal cycle differences in rat brain levels of pain-related cannabimimetic lipid mediators. Am J Physiol Regul Integr Comp Physiol 2006; 291: 349-358.
19. Ye Y, Lu Z, Zhou W. Pheromone effects on the human hypothalamus in relation to sexual orientation and gender. Handb Clin Neurol 2021; 182: 293-306.
20. Micevych PE, Meisel RL. Integrating neural circuits controlling female sexual behavior. Front Syst Neurosci 2017; 11: 42.
21. Clayton AH, Valladares-Juarez EM. Female sexual dysfunction. Med Clin North Am 2019; 103: 681-698.
22. Rastrelli G, Corona G, Maggi M. Testosterone and sexual function in men. Maturitas 2018; 112: 46-52.
23. Krysiak R, Okopień B. Niedobór androgenów u kobiet. Wiad Lek 2013; 66: 360-369.
24. Del Río JP, Molina S, Hidalgo-Lanussa O i wsp. Tibolone as hormonal therapy and neuroprotective agent. Trends Endocrinol Metab 2020; 31: 742-759.
25. Clements JN, Thompson B. Flibanserin for hypoactive sexual desire disorder in premenopausal women. JAAPA 2018; 31: 51-53.
26. Dhillon S, Keam SJ. Bremelanotide: first approval. Drugs 2019; 79: 1599-1606.
27. Segraves RT, Clayton A, Croft H i wsp. Bupropion sustained release for the treatment of hypoactive sexual desire disorder in premenopausal women. J Clin Psychopharmacol 2004; 24: 339-342.
28. Salerian AJ. Successful treatment of sexual dysfunction with dronabinol: a case report. J Clin Psychiatry 2004; 65: 1146-1147.
29. De Sousa SM, Chapman IM, Falhammar H i wsp. Dopa-testotoxicosis: disruptive hypersexuality in hypogonadal men with prolactinomas treated with dopamine agonists. Endocrine 2017; 55: 618-624.
30. Krysiak R, Kowalcze K, Okopień B. Sexual function and depressive symptoms in young women with hypoprolactinaemia. Clin Endocrinol (Oxf) 2020; 93: 482-488.
31. Krysiak R, Kowalcze K, Okopień B. Sexual function and depressive symptoms in men with hypoprolactinaemia secondary to overtreatment of prolactin excess: a pilot study. Endocrinol Diabetes Nutr (Engl Ed) 2021; S2530-0164(21): 00145-2.
32. Simonsen U, Comerma-Steffensen S, Andersson KE. Modulation of dopaminergic pathways to treat erectile dysfunction. Basic Clin Pharmacol Toxicol 2016; 119 (Suppl 3): 63-74.
33. Valevski A, Modai I, Zbarski E i wsp. Effect of amantadine on sexual dysfunction in neuroleptic-treated male schizophrenic patients. Clin Neuropharmacol 1998; 21: 355-357.
34. Krzystanek M, Warchala A, Trędzbor B i wsp. Amantadine in the treatment of sexual inactivity in schizophrenia patients taking atypical antipsychotics – the pilot case series study. Pharmaceuticals (Basel) 2021; 14: 947.
Tekst pochodzi z „Psychiatrii Spersonalizowanej” 1/2022. Czasopismo można zamówić na stronie: www.termedia.pl/ps/prenumerata.




