Nowe strategie terapii niedrobnokomórkowego raka płuca
U większości chorych NDRP rozpoznawany jest w zaawansowanym stadium (IIIB/IV), wymagającym systemowego leczenia. Rokowanie dla tych chorych jest nadal złe, 5 lat przeżywa w tej grupie tylko kilka procent chorych. Jedną z dotychczas stosowanych opcji terapeutycznych jest hamowanie receptora dla naskórkowego czynnika wzrostu (EGFR).
EGFR (HER1/ErbB1) był pierwszym opisanym receptorem z rodziny HER, obejmującej poza tym receptory HER2/neu (ErbB2), HER3 (ErbB3) i HER4 (ErbB4).
Ekspresja EGFR stwierdzana jest w kilku nowotworach (płuca, piersi, jelita grubego, przełyku), a często jest to nawet nadekspresja (np. w 40 – 80% NDRP). Liczne ligandy, łącząc się z poszczególnymi receptorami powodują ich dimeryzację, aktywującą proces transdukcji sygnału do wnętrza komórki. Może to być homodimeryzacja (np. EGFR-EGFR) lub heterodimeryzacja (np. EGFR-HER2). W tym drugim przypadku HER2 jest preferowanym partnerem do połączeń z innymi receptorami, a pary heterodimeryczne wywołują większy efekt mitogenny i przekaźnikowy.
Aktywacja EGFR prowadzi do nasilenia różnych funkcji komórki, w tym proliferacji, migracji i przeżycia (hamowanie apoptozy).
Mutacje w zakresie EGFR opisano po raz pierwszy u chorych z istotną odpowiedzią na gefitynib. Delecje, wymiany i inne rzadsze mutacje stwierdzane w domenie związanej z kinazą tyrozynową (eksony 18 – 21) mają charakter aktywujący i powodują, że funkcje kinazy pozostają aktywne nawet bez obecności liganda. Te mutacje aktywujące są ściśle związane z większą wrażliwością komórek na EGFR TKI. Stwierdzono również obecność mutacji powodujących oporność na inhibitory EGFR – opisano je w dalszej części.
Inhibitory EGFR
Nadekspresja EGFR, obecność aktywujących (uwrażliwiających na inhibitory) mutacji, istotna rola w biologii komórek guza spowodowały, że EGFR stał się celem dla leków z grupy terapii celowanych, do których należą drobnocząsteczkowe odwracalne inhibitory KT EGFR gefitynib i erlotynib oraz przeciwciało bezpośrednio skierowane przeciwko domenie zewnątrzkomórkowej receptora – cetuksymab.
Stosowanie odwracalnych inhibitorów KT EGFR u chorych z NDRP przynosi efekt kliniczny w pewnych populacjach, jednak skuteczność gefitynibu i erlotynibu w populacji ogólnej jest umiarkowana. Jak pokazują badania, odpowiedź guza uzyskuje się tylko u ok. 10% chorych nie podlegających wstępnej selekcji. Z analiz retrospektywnych wynika, że istnieją pewne cechy fenotypowe związane z większym prawdopodobieństwem odpowiedzi na odwracalne TKI EGFR: pochodzenie azjatyckie, płeć żeńska, brak palenia oraz histologia adenocarcinoma, niemniej jednak znaczenie tych cech jako czynników predykcyjnych pozostaje dyskusyjne. Zidentyfikowano kilka biomarkerów molekularnych, które mogą być czynnikami predykcyjnymi skuteczności TKI EGFR. Wydaje się, że najefektywniejszym jest obecność mutacji aktywujących w EGFR. Stwierdza się je u ok. 10% chorych razy kaukaskiej i u 25-50% chorych pochodzących z Azji i są one częściej związane z takimi cechami jak niepalenie, histologia adenocarcinoma oraz cechy bronchioalweolarne. Ok. 90% tych mutacji zlokalizowanych jest w eksonach 19 i 21. U chorych z mutacjami w eksonie 19 stwierdzono wyższy odsetek odpowiedzi (20-90% w porównaniu z 10-20% w populacji ogólnej) jak i dłuższe przeżycia całkowite po zastosowaniu TKI EGFR. Do innych biomarkerów należą: ocena liczby kopii genu w badaniu FISH (ang. fluorescencji in-situ hybridization), ekspresja receptora w badaniu immunohistochemicznym (IHC), mająca raczej mniejsze znaczenie oraz mutacje w genie KRAS.
Oporność na odwracalne TKI EGFR
Największym ograniczeniem stosowania gefitynibu i erlotynibu jest pojawiająca się oporność na te leki. Prawie u wszystkich chorych, którzy początkowo odpowiedzieli na gefitynib i erlotynib dochodzi w końcu do nawrotu choroby. Wynika to z faktu, że większość chorych otrzymujących te leki jest albo wyjściowo na nie oporna albo też oporność rozwija się w takcie terapii. Długość odpowiedzi na gefitynib lub erlotynib wynosiła w badaniach klinicznych 6 – 8 miesięcy, mimo iż u niektórych chorych odpowiedź trwała nawet kilka lat.
Mechanizmy oporności pierwotnej
1. Mutacje polegające na insercji w eksonie 20 EGFR, które obniżają wrażliwość na odwracalne TKI około 100-krotnie oraz mutacja w eksonie 20 – T790M, pojawiająca się w skojarzeniu z mutacjami aktywującymi EGFR.
2. Mutacje w genie KRAS, stwierdzane u ok. 15-30% chorych z NDRP. W kilku badaniach sugerowano, że mutacje KRAS, stwierdzane zwykle w kodonach 12 i 13 eksonu 2 oznaczają oporność pierwotną zarówno na geftynib jak i erlotynib, niemniej jednak w badaniu SATURN stwierdzono podobną korzyść kliniczną u chorych z mutacjami i wild-type KRAS.
3. Utrata białka PTEN (ang. phosphatase and tensin homologue), co skutkuje nadmierną aktywnością kinazy fosfatydyloinozytolu-3 (Pl3K) i serynowo – treoninowej kinazy Akt, promującą przeżycie komórek NDRP. Utrata białka PTEN wiąże się z opornością na gefitynib i erlotynib oraz gorszą prognozą.
4. Amplifikacja genu MET, receptora dla czynnika wzrostu hepatocytów, wiąże się z dodatkowym sygnałem ułatwiającym przeżycie komórek NDRP. Amplifikacja lub nadekspresja MET stwierdzana jest u ok. 3 – 7% chorych nie otrzymujących żadnych TKI EGFR.
Mechanizmy oporności nabytej
1. Wtórna mutacja T790M, pojawiająca się głównie w komórkach guza z pierwotną obecnością mutacji aktywujących EGFR, zwykle w eksonie 19 (L858R). Uważa się, że mutacja T790M pojawia się w trakcie terapii TKI EGFR, jednak wykazano też jej obecność w nawet 38% biopsji guza pochodzących od chorych nieleczonych TKI EGFR. T790M nie uniemożliwia odpowiedzi na gefitynib czy erlotynib, niemniej jednak PFS jest istotnie krótszy (7,7 vs. 16,5miesiąca; p<0,001). Uważa się, że mutacja T790M może być użytecznym biomarkerem, pozwalającym na identyfikacje chorych mających niewielkie prawdopodobieństwo uzyskania trwałej odpowiedzi.
2. Mutacja D761Y, związana z nabytą odpornością na gefitynib i erlotynib.
3. Wtórna mutacja T854A, powodującą utratę zdolności wiązania odwracalnych TKI EGFR.
4. Heterogenność genetyczna komórek NDRP może być również czynnikiem rozwoju oporności. W sytuacji, kiedy w guzie znajdują się komórki z mutacjami aktywującymi EGFR oraz z wild-type, terapia prowadzona z zastosowaniem TKI EGFR może doprowadzić do eliminacji klonów komórkowych z mutacjami i w konsekwencji do selekcji komórek z wild-type, opornych na TKI EGFR.
5. Amplifikacja MET, stwierdzana w ok. 20% przypadków nabytej oporności na gefitynib i erlotynib.
6. Zwiększona ekspresja receptora dla insulinopodobnego czynnika wzrostu-1 (IGF-1R, ang. insulin-like growth factor-1 receptor) i heterodimeryzacja z EGFR. Dostępne dane kliniczne wskazują jednak, że zwiększona ekspresja IGF-1R koreluje z lepszą odpowiedzią na gefitynib u chorych z NDRP, stąd konieczne są dalsze badania dla pełnego wyjaśnienia roli tego receptora.
7. Inne: zwiększona internalizacja EGFR, zwiększona ekspresja ludzkiego białka transportującego leki ABCG2.
Nieodwracalne inhibitory EGFR
Nieodwracalne inhibitory EGFR różnią się chemicznie od odwracalnych, przez co formują kowalentne połączenie z Cys-733 (w innym systemie numeracyjnym Cys-797). W związku z tym przełamanie działania hamującego receptor możliwe jest tylko na drodze syntezy nowych białek. Nieodwracalne inhibitory EGFR wykazują również aktywność w komórkach z obecnością mutacji T790M.
Dodatkową zaletą nieodwracalnych inhibitorów EGFR jest fakt, że hamują one równocześnie inne receptory, np. HER2 lub tez wszystkie receptory z rodziny HER (tzw. inhibitory pan-ErbB).
HER2 jest preferowanym partnerem w procesie dimeryzacji dla innych receptorów z rodziny HER, w tym EGFR. Sygnały mitotyczne przekazywane przez EGFR mogą być wzmocnione przez koekspresję z HER2. Wykazano, że wysoka jednoczesna koekspresja EGFR i HER2 jest związana u chorych z NDRP w stadium I do IIIA z niekorzystna prognozą. Taka jednoczesna koekspresja może powodować zwiększone tworzenie heterodiemrów EGFR/HER2, wynikiem czego jest bardziej agresywny fenotyp. Wykazano też, że amplifikacja HER2 u chorych z NDRP, szczególnie w połączeniu z mutacją lub amplifikacją/nadekspresją EGFR koreluje z wrażliwością na TKI EGFR.
BIBW2992 (afatynib)
Jest nieodwracalnym inhibitorem EGFR i HER2., wykazującym aktywność przeciwko wild-type oraz zmutowanym formom EGFR, w tym z obecnością T790M. Związek ten wykazał większy potencjał niż erlotynib, gefitynib i lapatynib w wywoływaniu śmierci komórek z linii NDRP, w tym komórek z wild-type EGFR, z aktywującą mutacją L858R oraz z mutacją T790M, warunkującą oporność na erlotynib. Dodatkowo afatynib wykazał aktywność przeciwko komórkom zawierającym inne mutacje, takie jak mutacja HER2 (776insV) czy wtórne mutacje EGFR (T854A, D770-771insNPG). W badaniach I fazy z monoterapią afatynibem wykazano, że działania niepożądane obejmują głównie biegunki i wysypki skórne, jednak okazały się one możliwe do opanowania. W tej fazie badań u 3 z 12 chorych z NDRP potwierdzono odpowiedź częściową, u 2 z nich stwierdzono mutacje del19. Badanie Lux-Lung 2 to badanie fazy II, obejmujące chorych z zaawansowanym rakiem gruczołowym płuca, z obecnością mutacji aktywujących EGFR, którzy nie byli wcześniej leczeni chemioterapią lub też byli po niepowodzeniu wcześniej zastosowanej chemioterapii. W grupie 67 chorych, którzy otrzymywali afatynib w drugiej linii leczenia odnotowano ORR (CR + PR) wynoszącą 64%, a odsetek kontroli choroby (DCR = ORR + SD) równy 96%. Mediana PFS wynosiła 10,2 miesiąca, a mediana okresu obserwacji 6,6 miesiąca. Badanie potwierdziło znany już profil bezpieczeństwa leku, najczęściej obserwowanymi działaniami niepożądanymi były zmiany skórne i biegunki. Obecnie trwa randomizowane badanie IIB/III fazy Lux-Lung 1, oceniające skuteczność afatynibu w porównaniu z placebo u chorych, u których doszło do niepowodzenia po wcześniejszej terapii erlotynibem lub gefitynibem. Z kolei w badaniu fazy III Lux-Lung 3, rozpoczynającym rekrutację, oceniana będzie skutecznośc afatynibu w porównaniu z chemioterapią cisplatyna/pemetreksed u chorych z NDRP i mutacjami EGFR w I linii terapii.
HKI-272 (neratynib)
Jest nieodwracalnym inhibitorem EGFR i HER2. W badaniach przedklinicznych wykazał większy niż gefitynib potencjał w hamowaniu indukowanej liganiem autofosforyzacji EGFR i przekazywaniu sygnału do komórki w liniach komórkowych z obecnością mutacji L858R jak i T790M. Jednakże wyniki aktualnych badań na liniach komórkowych wskazują, że przełamanie oporności związanej z mutacją T790M możliwe jest tylko przy stężeniach leku przekraczających te uzyskane w badaniach klinicznych. Neratynib wykazał również skuteczność w komórkach z obecnością różnych mutacji HER2. W badaniach fazy I w grupie 14 chorych z NDRP nie odnotowano żadnej PR, natomiast obserwowano trwającą powyżej 24 tygodni SD u 6 chorych, którzy mieli niepowodzenie po wcześniejszej terapii erlotynibem lub gefitynibem. Najczęstszymi działaniami niepożądanymi były biegunki, nudności, wymioty, zmęczenie i brak apetytu. Badanie fazy II obejmowało chorych z zaawansowanym NDRP, otrzymujących wcześniej do 3 linii chemioterapii. Chorych podzielono na 3 grupy: 1/ z mutacjami EGFR, po niepowodzeniu erlotynibu lub gefitynibu, 2/ bez mutacji i 3/ bez wcześniejszego leczenia TKI EGFR. Nie wykazano żadnych różnic pomiędzy grupami w zakresie RR (2%, 2%, 4%0, częstości SD (47%, 46%, 39%) i mediany PFS (11,6, 14,7 i 7,4 tygodni). Obecnie nie prowadzone SA badania neratynibu u chorych z NDRP.
PF00299804
Jest doustnym, nieodwracalnym inhibitorem pan-HER. Potencjał hamowania komórek z wild-type EGFR jest podobny do erlotynibu, gefitynibu i innych odwracalnych inhibitorów pan-HER. W badaniach przedklinicznych związek ten wykazał aktywność w komórkach zawierających częste mutacje EGFR oraz w komórkach z podwójnymi mutacjami L858R/T790M w istotnie niższych IC50 niż gefitynib. W badaniu fazy I u 42 chorych z NDRP wykazano akceptowalną toksyczność, a najczęstsze działania niepożądane obejmowały biegunkę i zmiany skórne. Spośród 29 chorych objętych oceną odpowiedzi guza, u 2 stwierdzono PR, a u 8 – SD. Żaden z chorych z mutacja T790M nie odpowiedział, ale należy zaznaczyć, że ich liczna była mała. Badanie fazy II objęło chorych z zaawansowanym NDRP, z wild-type KRAS, po niepowodzeniu 1 lub więcej linii chemioterapii oraz po wcześniejszym leczeniu erlotynibem (ramię A – adenocarcinoma, ramię B – nie-adenocarcinoma). Dotychczas włączono do badania 34 chorych, wśród 20 objętych wstępną analizą odpowiedzi guza stwierdzono niepotwierdzoną PR u 2 chorych, oraz 10 przypadków SD z medianą czasu trwania odpowiedzi 11,5 tygodnia. Najczęstsze działania niepożądane obejmowały zaburzenia skóry i przewodu pokarmowego. U 2 chorych odnotowano zator płucny 4. stopnia.
Jest nieodwracalnym inhibitorem EGFR i HER2. Badanie fazy I z eskalacją dawki przeprowadzono u Japończyków z zaawansowanymi nowotworami ze stwierdzoną nadekspresją EGFR. W grupie 15 chorych 10 miało NDRP. MTD wyniosła 35 mg, a DLT obejmowały śródmiąższową chorobę płuc 4. stopnia i biegunkę 3. stopnia. Związek wykazał aktywność u 2 chorych z NDRP, z obecnością mutacji EGFR i nabytą opornością na gefitynib.
CI-1033
Jest nieodwracalnym inhibitorem Pan-ErbB. Badanie fazy I obejmowało 32 chorych z guzami litymi opornymi na standardowa terapię. Lek podawano doustnie przez 14 kolejnych dni w 21-dniowych cyklach. DLT przy dawce 560 mg/d obejmowały biegunkę, zmiany skórne i zaburzenia apetytu. Nie odnotowano obiektywnych odpowiedzi, a SD stwierdzono u 6 chorych. W badaniu fazy II, otwartym, randomizowanym u chorych z zaawansowanym NDRP po niepowodzeniu chemioterapii z cispaltyną wykazano, że CI-1033 wykazuje umiarkowaną aktywność w NDRP. U 4/166 (2,5%) chorych doszło do PR, a 30/166 (19%) miało SD. Odsetek jednorocznych przeżyć mieścił się w przedziale 26 – 29%. Obecnie nie toczą się żadne inne badania tego związku u chorych z NDRP.
AV-412/MP-412
Jest nieodwracalnym inhibitorem EGFR i HER2. Obecnie związek badany jest w badaniu I fazy z eskalacją dawki u chorych z zaawansowanymi nowotworami.
Podsumowanie
Hamowanie EGFR ma już swoje miejsce w leczeniu NDRP. Jednakże odpowiedzi kliniczne nie są częste w ogólnej populacji, a u większości chorych odpowiadających na aktualnie dostępne preparaty hamujące EGFR dochodzi w końcu do nawrotu choroby. Badania przedkliniczne wykazały, że jednoczesne hamowanie kilku receptorów z rodziny HER może być skuteczniejsze niż hamowanie tylko EGFR. Obecnie w różnych fazach rozwoju klinicznego znajduje się kilka cząsteczek, hamujących równocześnie ERGFR/HER2 lub wszystkie receptory HER (pan-HER).
Nieodwracalne TKI są obiecującą, nową klasą leków, które mogą przełamać lub zapobiec pojawieniu się oporności na odwracalne TKI EGFR. Pozostaje do wyjaśnienia, czy zwiększone hamowanie EGFR przez tę nową generację inhibitorów jest efektem przełamania/zapobiegania oporności czy jest może efektem zwiększonej skuteczności, związanej z jednoczesnym hamowaniem EGFR i HER2.
Oczekiwane są wyniki badań fazy II i III tych związków u chorych z NDRP, szczególnie wśród opornych na odwracalne inhibitory EGFR.
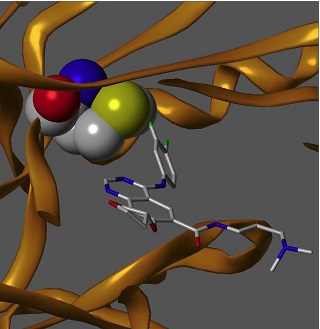
Met 790 - mutacja T790M uniemożliwiająca działanie odwracalnych inhibitorów kinazy tyrozynowej.
Thr 790
Ekspresja EGFR stwierdzana jest w kilku nowotworach (płuca, piersi, jelita grubego, przełyku), a często jest to nawet nadekspresja (np. w 40 – 80% NDRP). Liczne ligandy, łącząc się z poszczególnymi receptorami powodują ich dimeryzację, aktywującą proces transdukcji sygnału do wnętrza komórki. Może to być homodimeryzacja (np. EGFR-EGFR) lub heterodimeryzacja (np. EGFR-HER2). W tym drugim przypadku HER2 jest preferowanym partnerem do połączeń z innymi receptorami, a pary heterodimeryczne wywołują większy efekt mitogenny i przekaźnikowy.
Aktywacja EGFR prowadzi do nasilenia różnych funkcji komórki, w tym proliferacji, migracji i przeżycia (hamowanie apoptozy).
Mutacje w zakresie EGFR opisano po raz pierwszy u chorych z istotną odpowiedzią na gefitynib. Delecje, wymiany i inne rzadsze mutacje stwierdzane w domenie związanej z kinazą tyrozynową (eksony 18 – 21) mają charakter aktywujący i powodują, że funkcje kinazy pozostają aktywne nawet bez obecności liganda. Te mutacje aktywujące są ściśle związane z większą wrażliwością komórek na EGFR TKI. Stwierdzono również obecność mutacji powodujących oporność na inhibitory EGFR – opisano je w dalszej części.
Inhibitory EGFR
Nadekspresja EGFR, obecność aktywujących (uwrażliwiających na inhibitory) mutacji, istotna rola w biologii komórek guza spowodowały, że EGFR stał się celem dla leków z grupy terapii celowanych, do których należą drobnocząsteczkowe odwracalne inhibitory KT EGFR gefitynib i erlotynib oraz przeciwciało bezpośrednio skierowane przeciwko domenie zewnątrzkomórkowej receptora – cetuksymab.
Stosowanie odwracalnych inhibitorów KT EGFR u chorych z NDRP przynosi efekt kliniczny w pewnych populacjach, jednak skuteczność gefitynibu i erlotynibu w populacji ogólnej jest umiarkowana. Jak pokazują badania, odpowiedź guza uzyskuje się tylko u ok. 10% chorych nie podlegających wstępnej selekcji. Z analiz retrospektywnych wynika, że istnieją pewne cechy fenotypowe związane z większym prawdopodobieństwem odpowiedzi na odwracalne TKI EGFR: pochodzenie azjatyckie, płeć żeńska, brak palenia oraz histologia adenocarcinoma, niemniej jednak znaczenie tych cech jako czynników predykcyjnych pozostaje dyskusyjne. Zidentyfikowano kilka biomarkerów molekularnych, które mogą być czynnikami predykcyjnymi skuteczności TKI EGFR. Wydaje się, że najefektywniejszym jest obecność mutacji aktywujących w EGFR. Stwierdza się je u ok. 10% chorych razy kaukaskiej i u 25-50% chorych pochodzących z Azji i są one częściej związane z takimi cechami jak niepalenie, histologia adenocarcinoma oraz cechy bronchioalweolarne. Ok. 90% tych mutacji zlokalizowanych jest w eksonach 19 i 21. U chorych z mutacjami w eksonie 19 stwierdzono wyższy odsetek odpowiedzi (20-90% w porównaniu z 10-20% w populacji ogólnej) jak i dłuższe przeżycia całkowite po zastosowaniu TKI EGFR. Do innych biomarkerów należą: ocena liczby kopii genu w badaniu FISH (ang. fluorescencji in-situ hybridization), ekspresja receptora w badaniu immunohistochemicznym (IHC), mająca raczej mniejsze znaczenie oraz mutacje w genie KRAS.
Oporność na odwracalne TKI EGFR
Największym ograniczeniem stosowania gefitynibu i erlotynibu jest pojawiająca się oporność na te leki. Prawie u wszystkich chorych, którzy początkowo odpowiedzieli na gefitynib i erlotynib dochodzi w końcu do nawrotu choroby. Wynika to z faktu, że większość chorych otrzymujących te leki jest albo wyjściowo na nie oporna albo też oporność rozwija się w takcie terapii. Długość odpowiedzi na gefitynib lub erlotynib wynosiła w badaniach klinicznych 6 – 8 miesięcy, mimo iż u niektórych chorych odpowiedź trwała nawet kilka lat.
Mechanizmy oporności pierwotnej
1. Mutacje polegające na insercji w eksonie 20 EGFR, które obniżają wrażliwość na odwracalne TKI około 100-krotnie oraz mutacja w eksonie 20 – T790M, pojawiająca się w skojarzeniu z mutacjami aktywującymi EGFR.
2. Mutacje w genie KRAS, stwierdzane u ok. 15-30% chorych z NDRP. W kilku badaniach sugerowano, że mutacje KRAS, stwierdzane zwykle w kodonach 12 i 13 eksonu 2 oznaczają oporność pierwotną zarówno na geftynib jak i erlotynib, niemniej jednak w badaniu SATURN stwierdzono podobną korzyść kliniczną u chorych z mutacjami i wild-type KRAS.
3. Utrata białka PTEN (ang. phosphatase and tensin homologue), co skutkuje nadmierną aktywnością kinazy fosfatydyloinozytolu-3 (Pl3K) i serynowo – treoninowej kinazy Akt, promującą przeżycie komórek NDRP. Utrata białka PTEN wiąże się z opornością na gefitynib i erlotynib oraz gorszą prognozą.
4. Amplifikacja genu MET, receptora dla czynnika wzrostu hepatocytów, wiąże się z dodatkowym sygnałem ułatwiającym przeżycie komórek NDRP. Amplifikacja lub nadekspresja MET stwierdzana jest u ok. 3 – 7% chorych nie otrzymujących żadnych TKI EGFR.
Mechanizmy oporności nabytej
1. Wtórna mutacja T790M, pojawiająca się głównie w komórkach guza z pierwotną obecnością mutacji aktywujących EGFR, zwykle w eksonie 19 (L858R). Uważa się, że mutacja T790M pojawia się w trakcie terapii TKI EGFR, jednak wykazano też jej obecność w nawet 38% biopsji guza pochodzących od chorych nieleczonych TKI EGFR. T790M nie uniemożliwia odpowiedzi na gefitynib czy erlotynib, niemniej jednak PFS jest istotnie krótszy (7,7 vs. 16,5miesiąca; p<0,001). Uważa się, że mutacja T790M może być użytecznym biomarkerem, pozwalającym na identyfikacje chorych mających niewielkie prawdopodobieństwo uzyskania trwałej odpowiedzi.
2. Mutacja D761Y, związana z nabytą odpornością na gefitynib i erlotynib.
3. Wtórna mutacja T854A, powodującą utratę zdolności wiązania odwracalnych TKI EGFR.
4. Heterogenność genetyczna komórek NDRP może być również czynnikiem rozwoju oporności. W sytuacji, kiedy w guzie znajdują się komórki z mutacjami aktywującymi EGFR oraz z wild-type, terapia prowadzona z zastosowaniem TKI EGFR może doprowadzić do eliminacji klonów komórkowych z mutacjami i w konsekwencji do selekcji komórek z wild-type, opornych na TKI EGFR.
5. Amplifikacja MET, stwierdzana w ok. 20% przypadków nabytej oporności na gefitynib i erlotynib.
6. Zwiększona ekspresja receptora dla insulinopodobnego czynnika wzrostu-1 (IGF-1R, ang. insulin-like growth factor-1 receptor) i heterodimeryzacja z EGFR. Dostępne dane kliniczne wskazują jednak, że zwiększona ekspresja IGF-1R koreluje z lepszą odpowiedzią na gefitynib u chorych z NDRP, stąd konieczne są dalsze badania dla pełnego wyjaśnienia roli tego receptora.
7. Inne: zwiększona internalizacja EGFR, zwiększona ekspresja ludzkiego białka transportującego leki ABCG2.
Nieodwracalne inhibitory EGFR
Nieodwracalne inhibitory EGFR różnią się chemicznie od odwracalnych, przez co formują kowalentne połączenie z Cys-733 (w innym systemie numeracyjnym Cys-797). W związku z tym przełamanie działania hamującego receptor możliwe jest tylko na drodze syntezy nowych białek. Nieodwracalne inhibitory EGFR wykazują również aktywność w komórkach z obecnością mutacji T790M.
Dodatkową zaletą nieodwracalnych inhibitorów EGFR jest fakt, że hamują one równocześnie inne receptory, np. HER2 lub tez wszystkie receptory z rodziny HER (tzw. inhibitory pan-ErbB).
HER2 jest preferowanym partnerem w procesie dimeryzacji dla innych receptorów z rodziny HER, w tym EGFR. Sygnały mitotyczne przekazywane przez EGFR mogą być wzmocnione przez koekspresję z HER2. Wykazano, że wysoka jednoczesna koekspresja EGFR i HER2 jest związana u chorych z NDRP w stadium I do IIIA z niekorzystna prognozą. Taka jednoczesna koekspresja może powodować zwiększone tworzenie heterodiemrów EGFR/HER2, wynikiem czego jest bardziej agresywny fenotyp. Wykazano też, że amplifikacja HER2 u chorych z NDRP, szczególnie w połączeniu z mutacją lub amplifikacją/nadekspresją EGFR koreluje z wrażliwością na TKI EGFR.
BIBW2992 (afatynib)
Jest nieodwracalnym inhibitorem EGFR i HER2., wykazującym aktywność przeciwko wild-type oraz zmutowanym formom EGFR, w tym z obecnością T790M. Związek ten wykazał większy potencjał niż erlotynib, gefitynib i lapatynib w wywoływaniu śmierci komórek z linii NDRP, w tym komórek z wild-type EGFR, z aktywującą mutacją L858R oraz z mutacją T790M, warunkującą oporność na erlotynib. Dodatkowo afatynib wykazał aktywność przeciwko komórkom zawierającym inne mutacje, takie jak mutacja HER2 (776insV) czy wtórne mutacje EGFR (T854A, D770-771insNPG). W badaniach I fazy z monoterapią afatynibem wykazano, że działania niepożądane obejmują głównie biegunki i wysypki skórne, jednak okazały się one możliwe do opanowania. W tej fazie badań u 3 z 12 chorych z NDRP potwierdzono odpowiedź częściową, u 2 z nich stwierdzono mutacje del19. Badanie Lux-Lung 2 to badanie fazy II, obejmujące chorych z zaawansowanym rakiem gruczołowym płuca, z obecnością mutacji aktywujących EGFR, którzy nie byli wcześniej leczeni chemioterapią lub też byli po niepowodzeniu wcześniej zastosowanej chemioterapii. W grupie 67 chorych, którzy otrzymywali afatynib w drugiej linii leczenia odnotowano ORR (CR + PR) wynoszącą 64%, a odsetek kontroli choroby (DCR = ORR + SD) równy 96%. Mediana PFS wynosiła 10,2 miesiąca, a mediana okresu obserwacji 6,6 miesiąca. Badanie potwierdziło znany już profil bezpieczeństwa leku, najczęściej obserwowanymi działaniami niepożądanymi były zmiany skórne i biegunki. Obecnie trwa randomizowane badanie IIB/III fazy Lux-Lung 1, oceniające skuteczność afatynibu w porównaniu z placebo u chorych, u których doszło do niepowodzenia po wcześniejszej terapii erlotynibem lub gefitynibem. Z kolei w badaniu fazy III Lux-Lung 3, rozpoczynającym rekrutację, oceniana będzie skutecznośc afatynibu w porównaniu z chemioterapią cisplatyna/pemetreksed u chorych z NDRP i mutacjami EGFR w I linii terapii.
HKI-272 (neratynib)
Jest nieodwracalnym inhibitorem EGFR i HER2. W badaniach przedklinicznych wykazał większy niż gefitynib potencjał w hamowaniu indukowanej liganiem autofosforyzacji EGFR i przekazywaniu sygnału do komórki w liniach komórkowych z obecnością mutacji L858R jak i T790M. Jednakże wyniki aktualnych badań na liniach komórkowych wskazują, że przełamanie oporności związanej z mutacją T790M możliwe jest tylko przy stężeniach leku przekraczających te uzyskane w badaniach klinicznych. Neratynib wykazał również skuteczność w komórkach z obecnością różnych mutacji HER2. W badaniach fazy I w grupie 14 chorych z NDRP nie odnotowano żadnej PR, natomiast obserwowano trwającą powyżej 24 tygodni SD u 6 chorych, którzy mieli niepowodzenie po wcześniejszej terapii erlotynibem lub gefitynibem. Najczęstszymi działaniami niepożądanymi były biegunki, nudności, wymioty, zmęczenie i brak apetytu. Badanie fazy II obejmowało chorych z zaawansowanym NDRP, otrzymujących wcześniej do 3 linii chemioterapii. Chorych podzielono na 3 grupy: 1/ z mutacjami EGFR, po niepowodzeniu erlotynibu lub gefitynibu, 2/ bez mutacji i 3/ bez wcześniejszego leczenia TKI EGFR. Nie wykazano żadnych różnic pomiędzy grupami w zakresie RR (2%, 2%, 4%0, częstości SD (47%, 46%, 39%) i mediany PFS (11,6, 14,7 i 7,4 tygodni). Obecnie nie prowadzone SA badania neratynibu u chorych z NDRP.
PF00299804
Jest doustnym, nieodwracalnym inhibitorem pan-HER. Potencjał hamowania komórek z wild-type EGFR jest podobny do erlotynibu, gefitynibu i innych odwracalnych inhibitorów pan-HER. W badaniach przedklinicznych związek ten wykazał aktywność w komórkach zawierających częste mutacje EGFR oraz w komórkach z podwójnymi mutacjami L858R/T790M w istotnie niższych IC50 niż gefitynib. W badaniu fazy I u 42 chorych z NDRP wykazano akceptowalną toksyczność, a najczęstsze działania niepożądane obejmowały biegunkę i zmiany skórne. Spośród 29 chorych objętych oceną odpowiedzi guza, u 2 stwierdzono PR, a u 8 – SD. Żaden z chorych z mutacja T790M nie odpowiedział, ale należy zaznaczyć, że ich liczna była mała. Badanie fazy II objęło chorych z zaawansowanym NDRP, z wild-type KRAS, po niepowodzeniu 1 lub więcej linii chemioterapii oraz po wcześniejszym leczeniu erlotynibem (ramię A – adenocarcinoma, ramię B – nie-adenocarcinoma). Dotychczas włączono do badania 34 chorych, wśród 20 objętych wstępną analizą odpowiedzi guza stwierdzono niepotwierdzoną PR u 2 chorych, oraz 10 przypadków SD z medianą czasu trwania odpowiedzi 11,5 tygodnia. Najczęstsze działania niepożądane obejmowały zaburzenia skóry i przewodu pokarmowego. U 2 chorych odnotowano zator płucny 4. stopnia.
Jest nieodwracalnym inhibitorem EGFR i HER2. Badanie fazy I z eskalacją dawki przeprowadzono u Japończyków z zaawansowanymi nowotworami ze stwierdzoną nadekspresją EGFR. W grupie 15 chorych 10 miało NDRP. MTD wyniosła 35 mg, a DLT obejmowały śródmiąższową chorobę płuc 4. stopnia i biegunkę 3. stopnia. Związek wykazał aktywność u 2 chorych z NDRP, z obecnością mutacji EGFR i nabytą opornością na gefitynib.
CI-1033
Jest nieodwracalnym inhibitorem Pan-ErbB. Badanie fazy I obejmowało 32 chorych z guzami litymi opornymi na standardowa terapię. Lek podawano doustnie przez 14 kolejnych dni w 21-dniowych cyklach. DLT przy dawce 560 mg/d obejmowały biegunkę, zmiany skórne i zaburzenia apetytu. Nie odnotowano obiektywnych odpowiedzi, a SD stwierdzono u 6 chorych. W badaniu fazy II, otwartym, randomizowanym u chorych z zaawansowanym NDRP po niepowodzeniu chemioterapii z cispaltyną wykazano, że CI-1033 wykazuje umiarkowaną aktywność w NDRP. U 4/166 (2,5%) chorych doszło do PR, a 30/166 (19%) miało SD. Odsetek jednorocznych przeżyć mieścił się w przedziale 26 – 29%. Obecnie nie toczą się żadne inne badania tego związku u chorych z NDRP.
AV-412/MP-412
Jest nieodwracalnym inhibitorem EGFR i HER2. Obecnie związek badany jest w badaniu I fazy z eskalacją dawki u chorych z zaawansowanymi nowotworami.
Podsumowanie
Hamowanie EGFR ma już swoje miejsce w leczeniu NDRP. Jednakże odpowiedzi kliniczne nie są częste w ogólnej populacji, a u większości chorych odpowiadających na aktualnie dostępne preparaty hamujące EGFR dochodzi w końcu do nawrotu choroby. Badania przedkliniczne wykazały, że jednoczesne hamowanie kilku receptorów z rodziny HER może być skuteczniejsze niż hamowanie tylko EGFR. Obecnie w różnych fazach rozwoju klinicznego znajduje się kilka cząsteczek, hamujących równocześnie ERGFR/HER2 lub wszystkie receptory HER (pan-HER).
Nieodwracalne TKI są obiecującą, nową klasą leków, które mogą przełamać lub zapobiec pojawieniu się oporności na odwracalne TKI EGFR. Pozostaje do wyjaśnienia, czy zwiększone hamowanie EGFR przez tę nową generację inhibitorów jest efektem przełamania/zapobiegania oporności czy jest może efektem zwiększonej skuteczności, związanej z jednoczesnym hamowaniem EGFR i HER2.
Oczekiwane są wyniki badań fazy II i III tych związków u chorych z NDRP, szczególnie wśród opornych na odwracalne inhibitory EGFR.
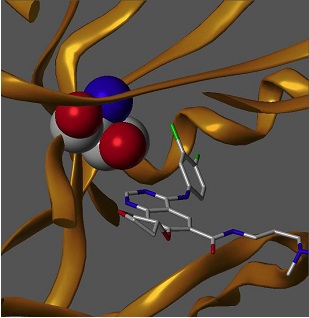
Met 790 - mutacja T790M uniemożliwiająca działanie odwracalnych inhibitorów kinazy tyrozynowej.
Thr 790
Źródło:
R. C. Doebele i wsp. New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer. Lung Cancer 2010; 69: 1-12, C. P. Belani The role of irreversible EGFR inhibitors in the treatment of non-small
R. C. Doebele i wsp. New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer. Lung Cancer 2010; 69: 1-12, C. P. Belani The role of irreversible EGFR inhibitors in the treatment of non-small
Kategorie:
Płuco i opłucna