
CASE DESCRIPTION
The case concerns a 61-year-old patient, referred to the clinic of neurology due to an incident of loss of consciousness of unknown aetiology. The patient was admitted to the clinic for the first time in February 2022. At that time he stated that the episode of loss of consciousness had occurred in October 2021 and did not reoccur. According to witnesses, the incident lasted a few minutes and was not accompanied by any additional symptoms, such as muscle contraction, tongue biting, seizure, involuntary urination or defecation. The patient had memory problems, feelings of slowness and impaired concentration abilities since the above-mentioned incident. According to his medical records, after the incident, the patient underwent neurological and cardiological diagnosis and levetiracetam 500 mg twice a day was then prescribed without the diagnosis of epilepsy. The electroencephalography (EEG) performed in October 2021 showed no seizure activity.
On admission to our clinic the patient presented history of untreated diabetes type II and hypertension. The patient neglected other chronic illnesses including cancer. The patient neglected pain, gastrointestinal issues and loss of weight. The first neurological examination at the clinic revealed no neurological deficits. The general examination revealed no pathology. Laboratory tests showed dyslipidemia, slight hyponatremia and slight hypochloremia. Other tests, such as blood count, kidney and liver function parameters, inflammatory parameters, coagulation factors and thyroid hormones showed no deviations from the normal values. An magnetic resonance imaging (MRI) with contrast of the head was performed and revealed an area of increased signal in the medial part of the left temporal lobe and numerous changes in the white matter considered to be vascular lesions in the fluid attenuated inversion recovery (FLAIR) sequence. A comparison was made with the previous examination from January 2022 and no changes in the image were found. During the first hospitalization, no seizure activity was recorded in the EEG examination.
In order to exclude other causes for loss of consciousness Doppler ultrasound examination of the carotid, vertebral and transcranial arteries was performed, and no hemodynamically significant changes were detected. Patients cognitive functions were assessed using the Montreal Cognitive Assessment Scale (MoCa) and were estimated 28/30 which is considered as a value within the normal range. During the conversation patient had visible trouble remembering words during delayed recall.
Based on a non-epileptic interview, physical examination and the results of additional tests, epilepsy was excluded and the treatment with levetiracetam was discontinued. Due to the low dose of levetiracetam a gradual decrease in dosage was not required.
The patient was discharged from the clinic with a recommendation for further diagnosis at the cardiology clinic for orthostatic causes of loss of consciousness. It was also recommended that the MRI of the head with contrast is repeated in 3 months time. Due to reported cognitive impairment, the patient was also referred to a psychological clinic.
After hospitalization, the patient experienced in May 2022 a new incident of loss of consciousness accompanied by rapid muscle contraction, involuntary urination and tongue biting. The interview suggested tonic-clonic seizures of focal onset (Classification by ILAE 2017). Additionally, the witnesses described short episodes of unresponsiveness of the patient when addressed. In May 2022, treatment with levetiracetam 500 mg twice a day was reinstated. Due to the occurrence of seizures and further memory disturbances, the patient was readmitted to the above-mentioned clinic of neurology in June 2022. The patient reported that his main complaint was increasing memory problems since the first loss of consciousness. During the neurological examination, the patient searched for words several times during the conversation and responded with delay. EEG (Figure I) and video EEG revealed moderate pathological changes in the temporal leads bilaterally (numeral series of theta waves with the frequency 5-7 Hz and sharp waves with the amplitude of 50-60 μV). Similar results were obtained in follow-up studies. The dose of levetiracetam was increased to 1000 mg twice a day. During the treatment with 1000 mg levetiracetam twice a day the measured level of levetiracetam in the blood serum was 9,80 μm/ml (normal values: 10,00-40,00 μm/ml). The final dose that provided satisfactory control of the seizures for the patient was levetiracetam 1500 mg twice a day. After an ophthalmological consultation, a lumbar puncture was performed. The cerebrospinal fluid analysis showed positive Pandy’s reaction and slightly elevated protein level (Table 1).
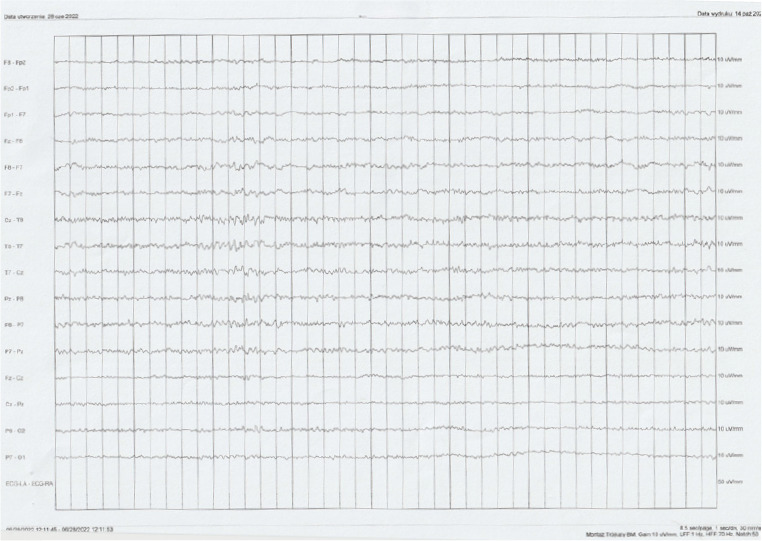
Table 1
Cerebrospinal fluid analysis
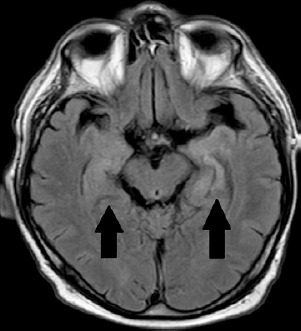
MRI of the head was repeated, which, similarly to the previous examination of June 2022, revealed a vaguely limited, poorly distinguished area of increased signal in the FLAIR sequence in the medial part of the left temporal lobe, without any signs of diffusion restriction or contrast enhancement. Temporal lobe dysplasia was initially suspected (Figure II). The patient also underwent psychological consultation 7th June 2022 – according to the MoCa, his result was 27/30. Comparing to the previous assessment there was a decline in the cognitive function. The declination was observed within the delayed recall.
In order to exclude other causes for loss of consciousness Holter ECG was performed and no cardiac arrhythmias were detected. Electroneurography and electromyography examination was performed, where the recording from the left rectus femoris muscle showed signs of chronic neurogenic damage with acute denervation in the form of fasciculation, fibrillation and positive sharp waves. Due to the subacute development of symptoms and presence of epileptic seizure accompanied by memory loss it was decided to expand the diagnosis to include autoimmune encephalitis and paraneoplastic syndromes. Laboratory tests showed an increase in the concentration of antibodies against leucine-rich glioma- inactivated 1 (LGI1) in the plasma serum (Table 2). The LGI1 antibodies were detected in the serum using immunofluorescence test kits with transfected cells.
Table 2
Antibodies associated with autoimmune encephalitis – diagnosis
| nMdA | Absent |
| CASpr2 | Absent |
| AMpA1-2 | Absent |
| LGI1 | Present |
| dppX | Absent |
| GABA | Absent |
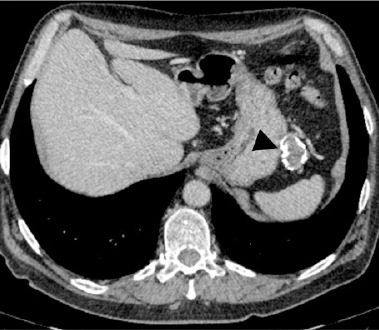
Due to the detection of antibodies against LGI1 CT scans of the chest, abdomen and pelvis were performed in order to exclude any underlying paraneoplastic process. The chest CT scan with contrast revealed no abnormalities. A CT scan of the abdomen and pelvis with contrast revealed a non-characteristic pathological mass between the stomach and spleen, measuring 45 × 35 × 27 mm with numerous calcifications, without any contrast enhancement (Figure III).
In order to examine the lesion more thoroughly, in accordance with the surgeon’s recommendation, the diagnosis was extended to include an MRI of the abdominal cavity with contrast and endosonography of the pancreas.An MRI examination of the abdominal cavity additionally showed that the lesion had a soft-tissue and fluid component. This lesion was partly adjacent to the stomach wall. The endoscopic ultrasound (EUS) examination did not locate the lesion. Gastrointestinal stromal tumour (GIST) was suspected. Based on the test results, the patient was diagnosed with autoimmune paraneoplastic encephalitis. The surgeon suggested a positron emission tomography scan and surgery as further steps.
During hospitalization, the patient had no epileptic seizures. At discharge, neurological examination revealed discrete asymmetry of deep reflexes. In the upper limbs the deep reflexes were more active on the right side, while in the lower limbs they were more active on the left side.
The patient’s cognitive functions were assessed 29th June 2022 to estimate the progression of symptoms. The patient received 26/30 points which showed another decline in the cognitive function. The patient showed progressive problems with delayed recall and word recognition.
The patient received at the beginning of July 2022 a seven doses of methylprednisolone with a mild, subjective improvement of cognitive functions.
In August 2022, the patient underwent the excision of the lesion. Intraoperatively, a tumour was found in a solid connection with a part of the stomach. There were no postoperative complications.
The pathological examination confirmed the diagnosis of GIST with the immunophenotype: CD117+, CD34+, CKAE1/AE3–, Desmin–, DOG1+, Ki-67–/+ in less than 1% of cells, S100–, SMA–. Category 1 according to AFIP. The tumour was completely removed.
After surgical treatment of the tumour, the patient’s condition significantly improved. A reduction in memory disorders and reduction of the frequency of epileptic seizures was also observed. Due to a mild presentation of symptoms the patient didn’t receive any immunomodulatory treatment after the operation. The level of antibodies was not assessed after the removal of the tumour in our clinic. Further follow-up was continued within a different clinic. We were provided information that a MRI scan was performed after the surgery. The sequences T1 and T2 revealed changes within the hippocamps described as post-inflammatory. An asymmetry of the hippocamps was observed – a reduction of the volume of the hippocamp on the left side was described.
DISCUSSION
Paraneoplastic neurological syndrome (PNS) is a clinical condition in which the functioning of the nervous system is impaired, not resulting from local tumour growth or metastases. The currently recognized underlaying mechanism of this condition is an autoimmune reaction occurring in the patient’s body. In response to the presence of tumour, antibodies called onconeural antibodies are produced, which are then directed against antigens present on cells of the nervous system [1, 2]. GIST is a diverse group of tumours with varying prognosis, with 30% being malignant [3]. The median age of diagnosis is between 66 and 69 years [4]. The specific precursor cells of GIST are investigated. Studies have shown that GISTs may originate from the pluripotent precursor cells of the interstitial cells of Cajal (ICC) [3, 5-8]. The origin of ICCs has remained unknown for many years because they have characteristics of both neuronal cells and connective tissue cells. Studies on animals have shown that they originate from the mesoderm. They are described as elongated, spindle-shaped cells with several projections. Cajal cells act as pacemakers in the digestive system. Just like the cells of the nervous system, they transmit and generate spontaneous, cyclic electrical impulses. These properties make ICCs form a network responsible for the coordinated contraction of the gastrointestinal muscles. Muscle cells, unlike Cajal cells, are not able to spontaneously generate nerve impulses. Cajal cells can form synapse-like connections with cells of the nervous system, thus playing an intermediary role between the nervous system and the muscles of the digestive system [9-11]. Analogously to the nervous system, Cajal cells express receptors for a number of mediators involved in neurotransmission, such as acetylcholine, substance P, nitric oxide, cyclic guanosine monophosphate and neurokinins. Receptors for serotonin 5-HT3 and 5-HT4 were also detected on their surface. The expression of receptors for vasoactive intestinal peptide, somatostatin, cholecystokinin A and bombesin as well as purinoreceptors were also found. The expression of individual receptors varies depending on the type of Cajal cell [12].
Cajal cells express the C-kit protein receptor (CD 117) encoded by the c-kit proto-oncogene. Mutation of the mentioned gene leads to the development of gastrointestinal stromal tumour [1, 3]. GIST tumours retain the expression of biomarkers present on Cajal cells [3]. GIST tumours were originally thought to originate from the autonomic nervous system of the gastrointestinal tract due to numerous features indicating this – projections, primitive intercellular connections and large cytoplasmic vacuoles. Further research revealed that the origin of this tumour is Cajal cells or their precursor cells [3, 7]. It can be assumed that the similarity in terms of surface receptors, function and structure of Cajal cells to cells of the nervous system may favour the development of the paraneoplastic neurological syndrome (PNS) in patients with GIST.
The patient met the diagnostic criteria for definite autoimmune limbic encephalitis (LE) constructed by Graus. He presented subacute progression of memory deficits and seizures. The MR showed lesions within the medial temporal lobes in the FLAIR sequence. The EEG showed pathological activity involving the temporal lobes. The cerebrospinal fluid (CSF) examination showed no pleocytosis. Alternative causes including infection were excluded [13].
Regarding to the diagnostic criteria of PNS definite limbic encephalitis is considered as a high-risk-phenotype. The detected anti-LG1 antibodies are considered as lower risk antibody for cancer. Considering the coincidence of detection of the tumour patient met the criteria of probable PNS [14]. The improvement after the resection of the tumour proves that the LE was most probably associated with the detected GIST tumour.
Anti-LGI1 anti-bodies are mentioned among the onconeural antibodies responsible for causing autoimmune encephalitis. They belong to antibodies directed against voltage-gated potassium channels (VGKC). LGI1 is a glycoprotein released by synaptic terminals to act on presynaptic ADAM metalloproteinases. It contributes to the inhibition of signal transduction through synapses. Anti-LGI1 encephalitis accounts for 11.2% of autoimmune encephalitis cases [13]. Symptoms include epileptic seizures. Their characteristic description can be found the literature. They usually affect patients between the fifth and eighth decade of life, but cases have also been reported between 31 and 84 years of age [2, 13]. Usually focal seizures occur, infrequently they are generalized, often recurrent and not easily treated [2, 13]. The pathognomonic symptom is faciobrachial dystonic seizures, occurring in approximately 40% of patients [2, 13]. These are short-term dystonic movements of the face and the upper limb on the same side, lasting several seconds. Sometimes they may also affect the lower limb [13]. During the diagnostic process it’s important to highlight that the anti-LGI1 antibody sensitivity in the blood is higher than CSF. That means it’s possible to receive negative anti-LGI1 antibody in the CSF while positive anti- LGI1 in the blood serum [16].
Epileptic seizures in autoimmune encephalitis, caused by anti-LGI1 antibodies, may be accompanied by cognitive disorders, including memory disorders, especially recent ones. They have an acute or subacute onset. Additionally, mental health disorders such as agitation, anxiety, behavioural changes, hallucinations and impulsive behaviour may occur. The occurrence of unexplained epileptic seizures combined with deterioration of cognitive functions should prompt further diagnosis [13-15]. Early treatment of anti-LGI1 encephalitis is crucial for the outcome and may prevent the development of dementia [16].
In 70% of patients, MRI of the brain shows changes with increased signal or signs of swelling in the temporal lobes, as was observed in this case (Figure II). Cerebrospinal fluid may be normal. Mild pleocytosis and increased protein levels are observed in 25% of patients [2].
An abnormality in laboratory tests occurring in approximately 60-70% of patients with limbic encephalitis is hyponatremia. This may be related to the expression of LGI1 in the kidneys and hypothalamus [2, 14]. It may be worth to mark that hyponatremia has been described as an isolated paraneoplastic syndrome associated with GIST tumour [20].
It can be observed that the symptoms of the patient from the described case in many respects coincided with the clinical signs of the disease described in the literature, with the exception of the fact that the patient did not present faciobrachial dystonic seizures. The patient’s age was within the established age range. The interview suggested tonic-clonic seizures of focal onset (Classification by ILAE 2017). The observed incidents of unresponsiveness may be classified as focal cognitive seizure with conduction aphasia. The inconvenience in diagnosing LE is the fact that the test results may be as in this case initially negative – the first EEG examination showed no pathological changes. The case showed us the importance of repeating the tests in case of susceptions of LE.
Paraneoplastic syndromes associated with GIST are rare. The described cases are very diverse and affect different organ systems of the body. In available sources we can find cases of hypoglycaemia [21-29], nephrotic syndrome [30, 31], hypothyroidism [32], hypocalcemia [33], hyponatremia [20], clubbed fingers and alopecia arealis [34] and myositis as a paraneoplastic syndromes associated with GIST [35]. A case of polymyalgia rheumatica has also been described as suspected to be another case of paraneoplastic syndrome [36] and one case of scleroderma-mimicking syndrome associated with gastrointestinal stromal tumour [37]. So far, only a few cases of PNS associated with GIST tumors have not been described in the literature. In available studies, we can find one case each of Guillain-Barré syndrome [38], severe parkinsonism [39], encephalitis [40] and headache with diplopia [41]. We can also find a described case of PNS presented as limbic encephalitis associated with GIST cancer with CV2/CRMP5 antibodies. The available literature does not provide any case of anti-LGI1-antibody autoimmune encephalitis as a PNS associated with GIST cancer. Among gastrointestinal cancers, the potential for PNS to occur has so far been demonstrated in patients with gastric cancer [42, 43]. Studies have shown that anti- VGKC antibodies may occur not only in cancer, but also in other gastrointestinal disorders such as irritable bowel syndrome or dyspepsia. However, the frequency did not differ from the case–control frequency and when detected, antibody levels were low. In some cases and controls, the detection of neural autoantibodies seemed to be predictive of neoplasia, but the predictive value of these tests needs further study [10].
The first-line therapy should be a surgical resection of the tumour causing PNS. Other treatment options include glucocorticoids, intravenous immunoglobulins (IVIG), plasmapheresis and immunosuppressive drugs such as azathioprine and rituximab [19]. Glucocorticoids have been shown to be more effective than IVIG [20]. However, the combination of glucocorticoid therapy and IVIG is more effective than glucocorticoid therapy alone [21]. In addition to treating the immune process itself, it is important to prevent epileptic seizures and treat other symptoms, such as changes in behaviour and mood. The treatment of epileptic seizures in patients with autoimmune encephalitis limited to antiepileptic drugs has a low effectiveness of up to 10% of cases. However, when combined with immunosuppressants, the effectiveness increases to 51% within 30 days and 88% within 90%. Moreover, there is an increased risk of cutaneous side effects of antiepileptic drugs in patients with anti-LGI1-associated encephalitis [2]. While treating we should take into consideration that many antiseizure drugs can cause drug-induced hyponatremia. This is important in case of LE where hyponatremia is commonly observed and an inappropriate choice of medication could dangerously lower the serum sodium level. Carbamazepine and oxcarbazepine are the anticonvulsants most commonly reported to be associated with hyponatremia in patients with epilepsy, although other anticonvulsants, such as eslicarbazepine, sodium valproate, lamotrigine, levetiracetam, and gabapentin, have also been reported to cause hyponatremia. Therefore carbamazepine and oxcarbazepine should be avoided in the treatment of LE [47, 48].
CONCLUSIONS
The case report presented in this paper proves the need for a thorough analysis of the cases of patients with newly developed memory disorders accompanied by epileptic seizures. What should draw our attention is the patient’s age, the dynamics of symptom development and the co-occurrence of epileptic seizures and memory disorders. Symptoms related to PNS may precede the detection of cancer by several months. Making the appropriate diagnosis is crucial for adjusting the treatment method and improving the patient’s health.
Based on the test results, the patient was diagnosed with autoimmune paraneoplastic encephalitis. After surgical treatment of cancer, the patient’s condition significantly improved with a reduction in memory disorders. The family claimed that the patient’s cognitive functions significantly improved and the seizures did not reoccur.






