
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a clonal neoplastic disorder of myeloid dendritic cells exhibiting a Langerhans cell phenotype, characterized by the expression of CD1a and CD207. The estimated annual incidence is 5–9 new cases per 1 million children and 1–2 new cases per 1 million adults [1]. The disease shows a peak incidence between 1 and 4 years of age, with infants younger than 1 year tending to present with more severe disease.
Current evidence suggests that LCH is associated with BRAF V600E mutations and other somatic mutations involving the mitogen-activated protein kinase kinase (MAP2K) pathway, resulting in increased activation of extracellular signal-regulated kinases (ERKs) within LCH cells [2, 3]. Clinically, LCH may present as single-system LCH (SS-LCH) or multisystem LCH (MS-LCH), involving 2 or more organ systems. Risk organ involvement includes the liver, spleen, and bone marrow. Consequently, the clinical spectrum ranges from localized involvement of a single bone to severe, widespread, and potentially life-threatening disease [4].
Given the rarity of LCH and the limited data available in the pediatric population of Eastern India, retrospective case series and meta-analyses play an important role in improving understanding of the disease and its clinical behavior.
OBJECTIVE
The aim of this study was to evaluate the clinical, epidemiological profile and treatment outcomes of children (< 18 years) with LCH.
MATERIAL AND METHODS
This was a retrospective analysis of 20 newly diagnosed children with LCH younger than 18 years. The study was conducted over a 3-year period from March 2022 to February 2025 at the outpatient department, inpatient department, and day care unit of the Department of Haematology, Nil Ratan Sircar Medical College and Hospital, Kolkata, India.
Informed consent was obtained in all cases. Diagnosis was based on clinical findings and histopathological and immunohistochemical examinations. A definitive diagnosis required positive immunostaining of atypical large cells with pale cytoplasm and reniform nuclei for CD1a and/or Langerin (CD207) [5]. Each patient underwent a comprehensive evaluation. The variables analyzed included age, sex, detailed medical history, and clinical findings based on cutaneous and systemic examinations.
Additional investigations included complete blood count, liver and renal function tests, serum electrolytes, coagulation profile, skeletal survey, chest radiography, positron emission tomography–computed tomography, lactate dehydrogenase levels, abdominal ultrasonography, skin biopsy, and immunohistochemical analysis for CD1a and S100 protein.
All patients were treated according to the Histiocyte Society Evaluation and Treatment Guidelines for LCH. Initial therapy consisted of a 6-week course of vinblastine (6 mg/m² per week) and prednisolone (40 mg/m² per day), with further treatment determined by therapeutic response [6]. Patients were followed up for a period of 1 year. Statistical analysis was performed using Statistical Package for the Social Sciences software, version 20.0.
RESULTS
The median age was 3 years (range: 10 months - 14 years), and the male-to-female ratio was 3 : 1. The majority of patients were younger than 5 years (65%, n = 13), followed by those aged 5–10 years (25%, n = 5) and 10–18 years (10%, n = 2). Failure to thrive was the most common presenting complaint (70%, n = 14).
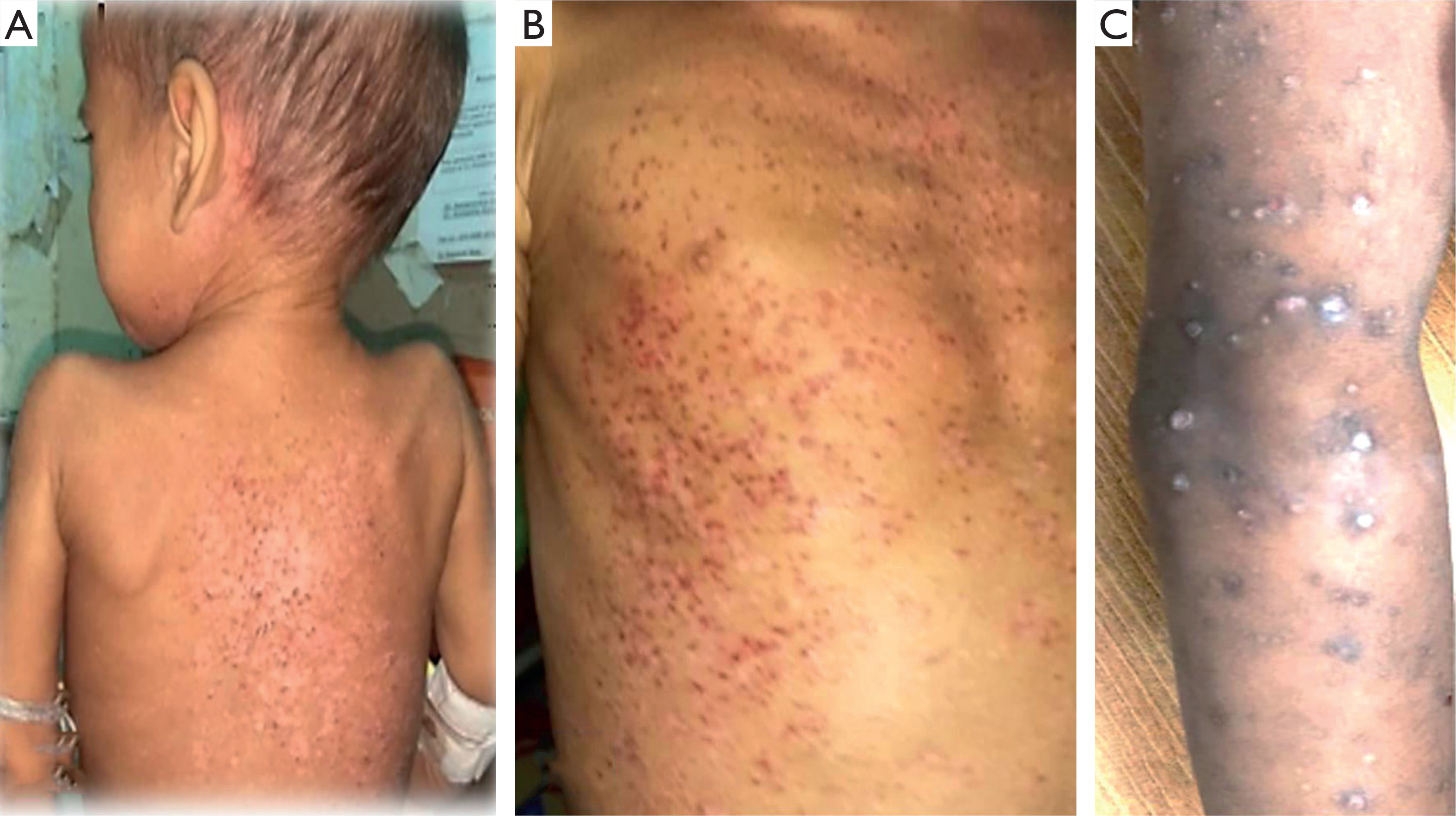
Multisystem involvement was observed in 75% of patients (n = 15), while 25% (n = 5) had SS-LCH. Skeletal involvement was present in all cases, and cutaneous involvement was noted in 50% (n = 10). Cutaneous lesions most commonly presented as red-brown scaly papules (50%), followed by seborrheic dermatitis-like lesions (30%), vesiculopustular lesions, and nodules (10% each). Cutaneous manifestations were more frequent in boys (70%) and most commonly involved the trunk, back, abdomen, and arms (Figure 1).
Figure 1
A child with Langerhans cell histiocytosis showing multiple brown maculopapular skin lesions with crusting involving the back (A), trunk (B), and arm (C)
Among patients with MS-LCH, risk organ involvement was identified in 86.6% of cases. The liver was the most frequently involved organ (73.3%), followed by the spleen (50%). Lymphadenopathy was present in 30% of patients, and bone marrow involvement was observed in 20%. Special site involvement included the ear (15%) and the central nervous system (10%).
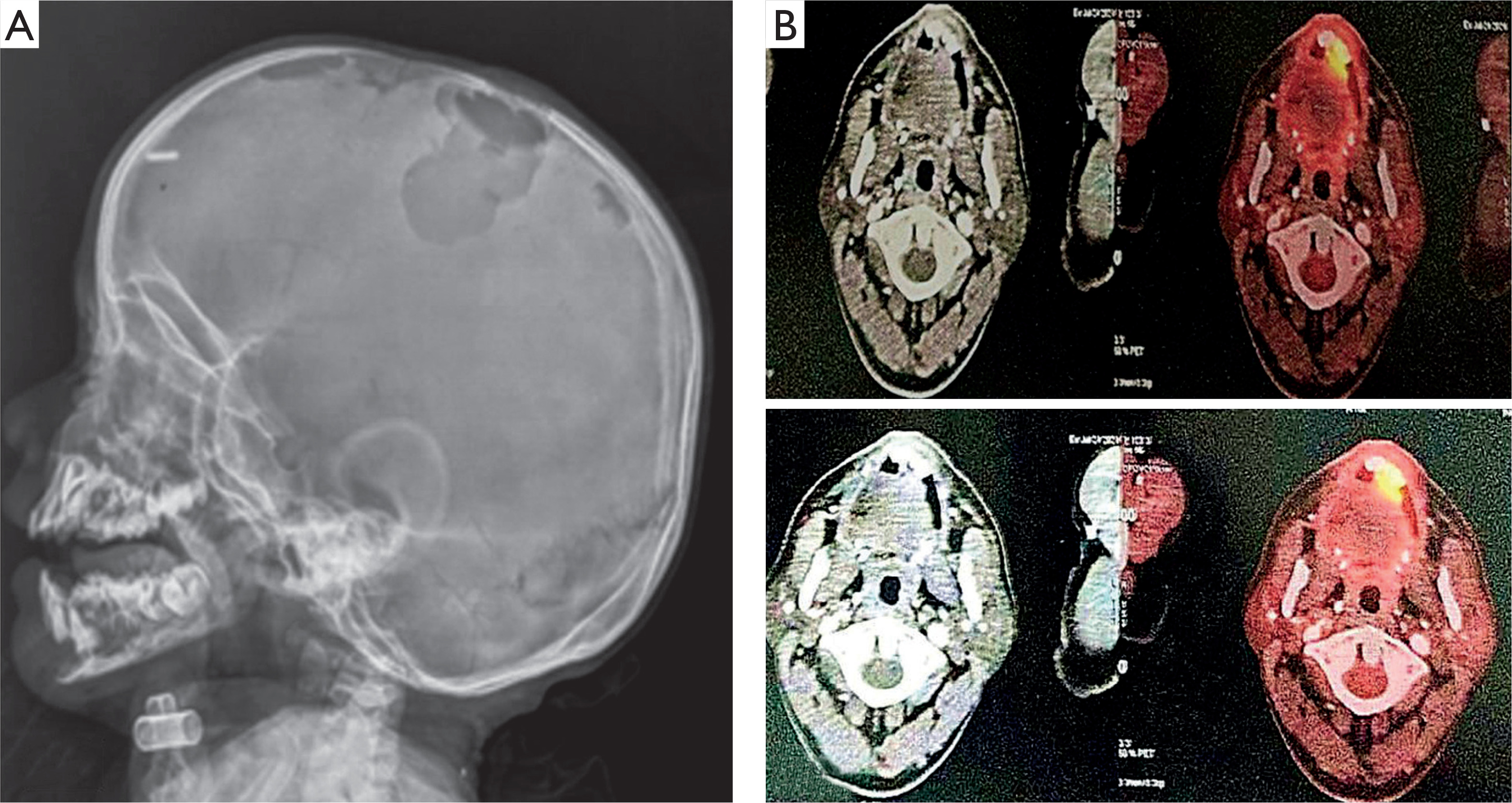
Laboratory evaluation revealed leukopenia in 15% of patients, anemia and thrombocytopenia in 20%, elevated alkaline phosphatase levels in 50%, and transaminitis in 35%. Skull X-ray demonstrated multiple lytic lesions, and positron emission tomography–computed tomography revealed metabolically active lytic lesions involving the skull and long bones in 75% of patients (Figure 2).
Figure 2
Lateral skull X-ray (A) showing multiple punched-out osteolytic lesions with well-defined, non-sclerotic margins involving both the outer and inner tables. Positron emission tomography-computed tomography scan (B) demonstrating multiple metabolically active lytic lesions involving the right maxilla and mandible, with associated soft tissue thickening in the right upper alveolus
After 6 weeks of therapy with vinblastine and prednisolone, 80% of patients achieved nonactive disease, while 20% had active disease. Among those with active disease, 75% showed partial response and 25% had progressive disease. Patients with nonactive disease continued maintenance therapy, those with partial response received an additional course of vinblastine and prednisolone, and 1 patient with progressive disease received salvage therapy with cytarabine and cladribine. One-year overall survival was 100%. A summary of findings is presented in Table 1.
Table 1
Summary of demographic, clinical, laboratory, radiological, and treatment-related findings in 20 paediatric patients with Langerhans cell histiocytosis
After 6 weeks of therapy with vinblastine and prednisolone, 80% of patients achieved nonactive disease, while 20% had active disease. Among those with active disease, 75% showed partial response and 25% had progressive disease. Patients with nonactive disease continued maintenance therapy, those with partial response received an additional course of vinblastine and prednisolone, and 1 patient with progressive disease received salvage therapy with cytarabine and cladribine. One-year overall survival was 100%.
A summary of demographic, clinical, laboratory, radiological, and treatment-related findings in 20 paediatric patients with LCH is presented in Table 1.
DISCUSSION
This study highlights the clinical, epidemiological profile, laboratory characteristics, and treatment outcomes of 20 children with LCH treated at a tertiary care center. The median age at presentation was 3 years, with a clear male predominance, which is consistent with findings reported by Alston et al. [7] and Guyot-Goubin et al. [8]. Similar demographic trends have also been reported by Dhar et al. [9] and Uppal et al. [10].
Clinical features
Failure to thrive and multisystem involvement were observed in the majority of patients in the present study, consistent with findings reported by Alston et al. [7] and Guyot-Goubin et al. [8]. In contrast, single-system involvement was more common in the study by Dhar et al. [9]. Skeletal involvement was observed in all patients, in keeping with existing literature identifying bone as the most frequently affected organ in LCH. Alston et al. [7] and Guyot-Goubin et al. [8] reported skeletal involvement in 67% and 75% of their cohorts, respectively. Conversely, in infants younger than 1 year, cutaneous involvement tends to be more prominent [11].
In the present study, cutaneous involvement was noted in 50% of cases, predominantly in boys, and most commonly presented as red scaly papules, followed by seborrheic dermatitis–like lesions. Similar findings were reported by Ng et al. [12], who identified folliculitis and red scaly papules as the most frequent cutaneous manifestations. The scalp was the most commonly affected site, observed in 7 of 10 patients, followed by the trunk in 4 of 10 patients.
Widodo et al. [13], in their case series of 20 patients with LCH, reported cutaneous involvement in 50% of cases with a male predominance. The most frequent clinical presentations included diffuse erythematous maculopapular eruptions, followed by seborrheic dermatitis–like lesions distributed across the scalp, face, trunk, groin, and extremities. Han et al. [14] similarly reported maculopapular lesions as the most common cutaneous manifestation and emphasized that LCH-related skin lesions may mimic eczema, viral exanthems, herpes infections, or other inflammatory dermatoses, often leading to misdiagnosis.
Multisystem disease with risk organ involvement, particularly affecting the liver and spleen, was identified in a substantial proportion of patients in the current study. These findings are comparable to those reported by Guyot-Goubin et al. [8], who observed that among 73 cases of disseminated LCH, the most frequently involved organs included the lungs (20 lungs), bone marrow with cytopenias (13 cases), and the liver or spleen (11 cases).
Laboratory findings
Hematological abnormalities and elevated liver enzyme levels were frequently observed. Similar findings have been reported by Mondal et al. [15], who documented anemia in a substantial proportion of patients. Transaminitis and increased uptake on positron emission tomography–computed tomography involving the skull and long bones were also observed.
Treatment
Initial therapy with vinblastine and prednisolone was effective in the majority of cases, with 80% of patients achieving nonactive disease at 6 weeks. One patient required salvage therapy with cladribine and cytarabine due to disease progression. The observed 1-year overall survival rate was 100% in the present study, which compares favourably with previous reports by Guyot-Goubin et al. [8] and Dhar et al. [9], who reported 1-year overall survival rates of 94% and 92%, respectively. These findings emphasize the importance of early diagnosis and adherence to standardized treatment protocols.
Among the limitations of the present study are the small sample size and the relatively short duration of follow-up. A comparison of the present study with previously published reports is summarized in Table 2.
Table 2
Comparison of the results/outcome of the present study with different other studies published in the literature
| Study | Number of patients | Median age [years] | Multisystem/Single system (%) | Skeletal/Cutaneous involvement (%) | One-year overall survival (%) |
|---|---|---|---|---|---|
| Alston et al. [7] | 101 | 2 | 56/44 | 67/37 | 74 |
| Guyot-Goubin et al. [8] | 251 | 3.5 | 43/57 | 75/34 | 94 |
| Dhar et al. [9] | 126 | 3 | 32/68 | 23/68 | 92 |
| Present study | 16 | 3 | 81/19 | 100/50 | 100 |
CONCLUSIONS
Langerhans cell histiocytosis is a rare, distinct, and highly heterogeneous condition with a wide spectrum of clinical manifestations. Early diagnosis is crucial for timely initiation of treatment and prevention of disease progression. Initial therapy with vinblastine and prednisolone is an effective treatment option, leading to favorable response rates and improved overall survival. Long-term follow-up remains essential to evaluate disease outcomes, including spontaneous resolution, organ system involvement, and mortality.










