
INTRODUCTION
Lesions that follow the lines of Blaschko represent a diagnostically challenging group of cutaneous mosaic disorders. Among them, hypomelanosis of Ito is a well-recognised entity, often classified as a neurocutaneous syndrome due to its frequent association with extracutaneous manifestations. In contrast, cases with identical skin morphology but without systemic involvement are often referred to as cutaneous-only forms of hypomelanosis of Ito, pigmentary mosaicism of the Ito type, or simply Blaschko-linear hypopigmentation [1–3].
The diagnosis of hypomelanosis of Ito is primarily clinical and relies on the recognition of characteristic cutaneous features following the lines of Blaschko. Comprehensive systemic evaluation is essential to identify or exclude associated extracutaneous manifestations. Although genetic testing and other supportive assessments may aid in the diagnostic process, their diagnostic value is limited by the mosaic nature of the condition.
OBJECTIVE
To present and analyse two paediatric patients with linear hypopigmentation following the lines of Blaschko – one with systemic involvement (diagnosed with hypomelanosis of Ito) and one with skin-only findings.
CASE REPORTS
Case 1
A 5-year-old boy was referred to the dermatology clinic due to the presence of hypopigmented macules, first noticed by his parents at the age of 2. The lesions followed the lines of Blaschko on the trunk and extremities (Figures 1 A, B). Physical examination revealed additional dysmorphic features, such as an elongated torso, a chest wall deformity consistent with pectus excavatum, a prominent abdomen, and an umbilical hernia. Early developmental history included difficulties with adaptation and mild developmental delays. Over time, the child’s development gradually reached age-appropriate milestones, and additional support was no longer required.
Figure 1
Case 1. A 5-year-old boy with hypopigmented streaks along Blaschko’s lines on the trunk and upper limbs (A, B)
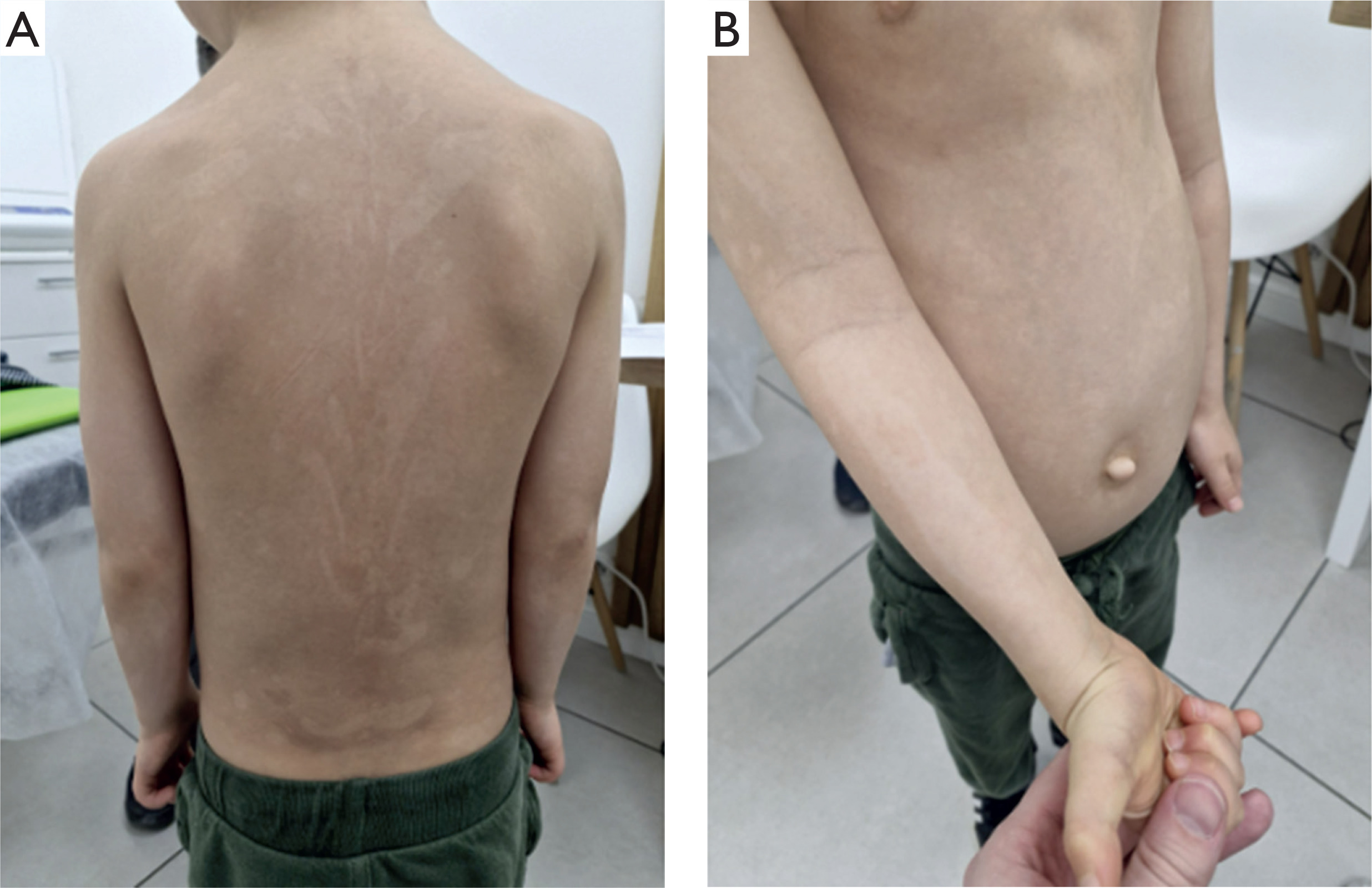
The patient remains under the care of several medical specialists due to ongoing clinical concerns. These include an otolaryngologist (for conductive hearing loss and recurrent otitis media with effusion), a cardiologist (due to a heart murmur and the presence of a fibrous chord), an ophthalmologist (for strabismus and amblyopia of the left eye), a pulmonologist (for chronic bronchitis with bronchiectasis), an orthopaedist (for spinal discomfort), and a posture clinic (for pectus excavatum).
Neurological assessment revealed no abnormalities, and the patient continues to be monitored as part of routine follow-up. Baseline laboratory investigations, including complete blood count, inflammatory markers, and metabolic panel, were within normal limits.
Genetic testing began with high-resolution karyotyping, which did not reveal any abnormalities. Subsequently, whole exome sequencing (WES) was performed. Although no pathogenic or likely pathogenic variants were identified which would explain the clinical features consistent with hypomelanosis of Ito, two notable findings emerged: a pathogenic heterozygous variant in the GJB2 gene (c.35del p.Gly12ValfsTer2), which is a well-known cause of autosomal recessive sensorineural hearing loss, and a variant of uncertain significance in the ORAI1 gene. The GJB2 variant may account for the patient’s auditory symptoms (including conductive hearing loss and recurrent otitis media with effusion); however, it does not explain the broader clinical phenotype and is not considered causative of the multisystem findings.
Based on the presence of pigmentary mosaicism, dysmorphic features, and multisystemic involvement, a clinical diagnosis of hypomelanosis of Ito was established by the clinical genetics team. The patient was advised to continue multidisciplinary specialist follow-up within a coordinated care framework.
Case 2
A 9-year-old boy was referred to the dermatology clinic for evaluation of hypopigmented macules distributed along the lines of Blaschko, affecting the trunk and extremities. The lesions fluoresced under Wood’s lamp, supporting the diagnosis of a pigmentary disturbance (Figures 2 A–E). According to the parents, the lesions had been present since early childhood and remained stable over time. On physical examination, the patient exhibited no dysmorphic features or neurological abnormalities.
Figure 2
Case 2. A 9-year-old boy with hypopigmented macules along Blaschko’s lines on the upper limbs and trunk visible indaylight (A–C) and under Wood’s lamp (D–F)
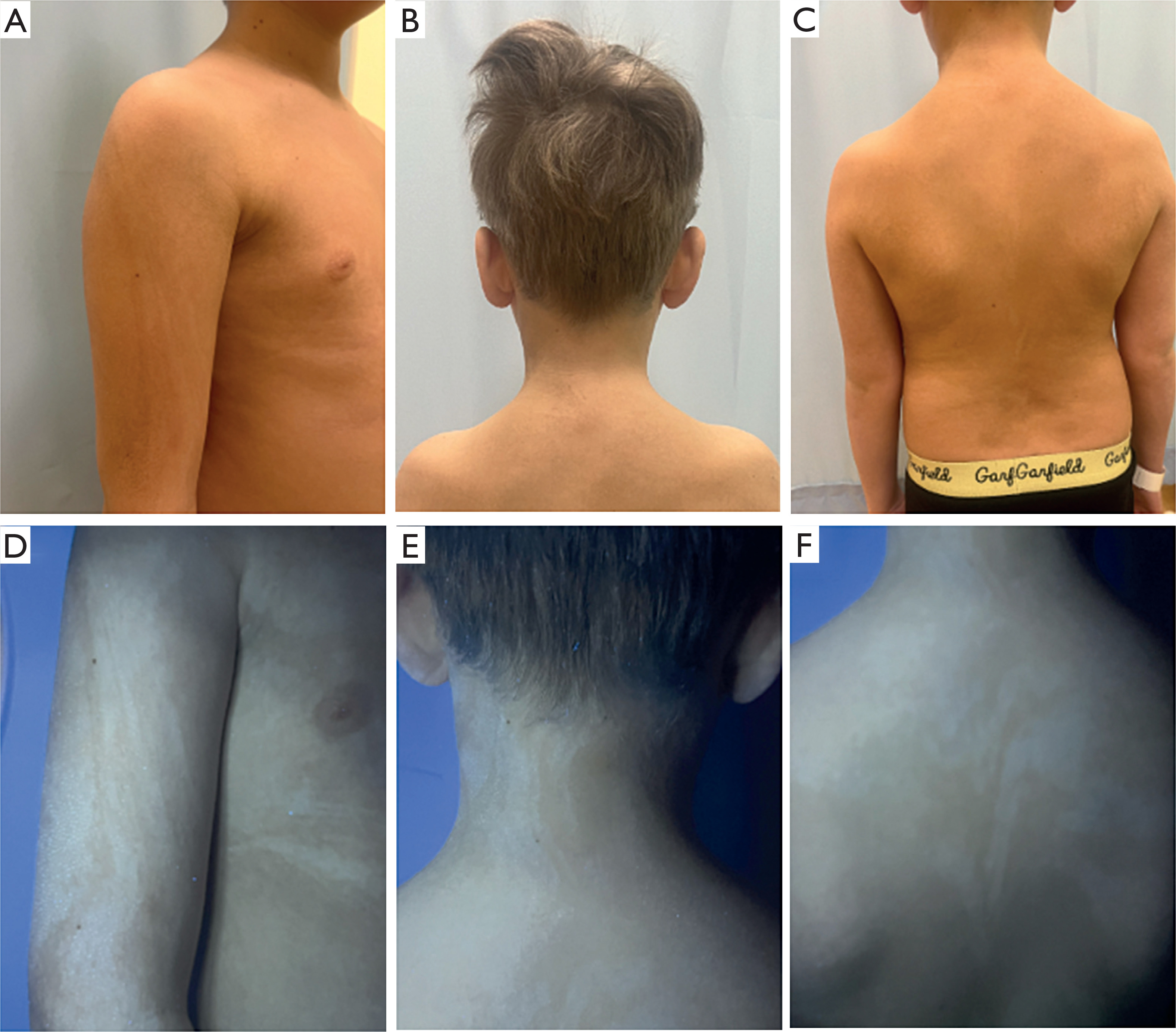
The child’s psychomotor development was within normal limits, and his medical history was unremarkable. As part of the initial diagnostic evaluation, baseline assessments were conducted to identify potential extracutaneous involvement. Neurological examination was normal. Cardiology consultation, including echocardiography, revealed no structural abnormalities. Ophthalmologic and otolaryngologic evaluations were also within normal limits, with no reported visual or auditory deficits. Routine laboratory tests, including screening for metabolic or inflammatory abnormalities, showed no significant abnormalities from reference values. The patient was not under the care of any other medical specialists apart from the dermatologist.
Given the absence of extracutaneous manifestations and the clinically stable cutaneous presentation, no advanced imaging or genetic testing were performed. The findings were considered consistent with pigmentary mosaicism of the Ito type. The patient remained under dermatologic follow-up, with recommendations for periodic reassessment in case extracutaneous features emerge over time.
DISCUSSION
Hypomelanosis of Ito is a condition characterized by hypopigmented lesions distributed along the lines of Blaschko. These lines are thought to reflect embryonic cell migration and serve as a visible sign of genetic mosaicism. This results from early postzygotic mutations that give rise to genetically distinct cell populations within the skin during embryonic development [2].
Clinically, it presents as macular areas of hypopigmentation arranged in characteristic whorls, streaks, and patches, which may appear unilaterally or bilaterally and are not preceded by inflammation. The distribution follows the Blaschko’s lines: ‘S’-shaped on the trunk, ‘V’-shaped on the back, linear on the limbs, and wavy on the scalp – patterns believed to reflect the underlying embryological migration of melanocytes [4]. These lesions may be congenital or manifest later in childhood, particularly in persons with lighter skin tones. The clinical phenotype is heterogeneous and depends on the type and extent of the underlying genetic mosaicism [3].
Minoru Ito’s first case involved a 22-year-old female patient with skin lesions that were limited only to the cutaneous surface, without systemic involvement [5]. Subsequently it became evident that, in certain cases, skin lesions could be associated with other developmental anomalies affecting internal organs [6]. Due to the frequent coexistence of neurological findings – particularly intellectual disability and epilepsy – hypomelanosis of Ito is now classified among the neurocutaneous syndromes. In terms of prevalence, it is considered the third most common disorder in this group, after neurofibromatosis and tuberous sclerosis complex [7–9].
The diagnosis of hypomelanosis of Ito is primarily clinical and relies on the identification of characteristic skin lesions in association with extracutaneous findings. Examination with a Wood’s lamp may enhance the visualization of hypopigmented lesions along the lines of Blaschko [10]. Once suspected, all patients should undergo comprehensive systemic evaluation, including neurological, musculoskeletal, cardiac, ophthalmologic, and otolaryngologic assessments. Additional evaluations such as renal ultrasound, developmental screening, and genetic consultation may be recommended based on clinical findings. Numerous associated clinical features have been documented in the literature, including mental and motor retardation, hypotonia, hyperkinesia, strabismus, ataxia, epilepsy, impaired or delayed speech, facial and limb asymmetry, and kyphoscoliosis [7, 11, 12]. Skeletal radiographs may be considered based on clinical findings suggestive of musculoskeletal abnormalities. Neuroimaging, such as computed tomography (CT) or magnetic resonance imaging (MRI), is indicated for individuals exhibiting neurological signs. Electroencephalography (EEG) should be performed in patients with seizures [8].
The diagnostic criteria established in 1992 by Ruiz-Maldonado et al. require one major or two minor findings in addition to characteristic cutaneous features. Major criteria include linear or whorled hypopigmented streaks involving more than two body segments, usually present from birth, along with at least one neurological or musculoskeletal abnormality. Minor criteria include chromosomal anomalies and the presence of at least two congenital malformations unrelated to the nervous or musculoskeletal systems. A definitive diagnosis is made when either one major plus one minor, or two minor criteria are fulfilled [7].
Although the diagnosis remains primarily clinical, genetic testing may assist in confirmation. Pathogenic variants or chromosomal anomalies can be detected using high-resolution karyotyping, chromosomal microarray, or next-generation sequencing. However, in mosaic disorders with a Blaschko-linear distribution, blood-based testing is frequently insufficient. This is because mosaicism may be confined to specific cell populations – particularly epidermal melanocytes – while being absent in peripheral blood or fibroblast-derived DNA. In such cases, a skin biopsy from the affected area may be necessary to obtain melanocytes for targeted analysis, often requiring epidermal cell separation techniques. These methods offer better diagnostic accuracy in mosaic disorders. Still, they are rarely used in everyday clinical settings due to technical and logistical barriers [3, 13–15]. Integrating these advanced approaches into clinical protocols may help address the diagnostic gap in suspected mosaic disorders.
Before establishing a diagnosis of hypomelanosis of Ito, it is essential to consider and exclude several pigmentary disorders that may present with a similar linear or whorled distribution of hypopigmentation along Blaschko’s lines. The three main entities to be ruled out are incontinentia pigmenti, particularly in its late hypopigmented stage; linear and whorled naevoid hypermelanosis; and systematized naevus depigmentosus [14].
In addition to these, the differential diagnosis should also consider a broader spectrum of disorders that may present with linear or patchy hypopigmentation. These include segmental vitiligo, various neurocutaneous syndromes (such as tuberous sclerosis or neurofibromatosis), and genetic conditions like Waardenburg syndrome and Tietz syndrome, both of which can involve pigmentary abnormalities in conjunction with systemic features such as sensorineural hearing loss or ocular defects [8, 14]. Careful clinical evaluation, including assessment of systemic involvement and family history, is essential to distinguish hypomelanosis of Ito from these overlapping disorders.
The treatment is primarily symptomatic and requires a multidisciplinary approach involving dermatologists, neurologists, paediatricians, orthopaedic surgeons, ophthalmologists, and geneticists. The skin lesions require no specific interventions. Certain regions of hypopigmentation may be covered up using makeup. Patients should be monitored for complications affecting any system with documented abnormalities. As part of the patient’s overall care, genetic counselling should be offered to family members [8].
Prognosis varies significantly depending on the presence and severity of extracutaneous involvement. Individuals with isolated skin findings typically have normal development and an excellent prognosis, whereas those with central nervous system abnormalities may face lifelong neurological and functional challenges [16].
CONCLUSIONS
These cases underscore the importance of comprehensive evaluation in individuals with pigmentary mosaicism. They also support a spectrum-based view of hypomelanosis of Ito, which can range from skin-only involvement to multisystemic disease. Proper diagnosis and long-term care require integration of dermatologic, neurological, and genetic perspectives. Ongoing clinical monitoring and interdisciplinary collaboration are crucial for optimizing outcomes in affected individuals.










