
SMA – choroba rzadka, dziedziczna

Rdzeniowy zanik mięśni (SMA, ang. spinal muscular atrophy) jest ciężką chorobą rzadką, prowadzącą do nieodwracalnego uszkodzenia neuronów ruchowych, odpowiedzialnych za właściwe funkcjonowanie mięśni szkieletowych, co prowadzi do ich postępującego osłabienia, a w konsekwencji zaniku.
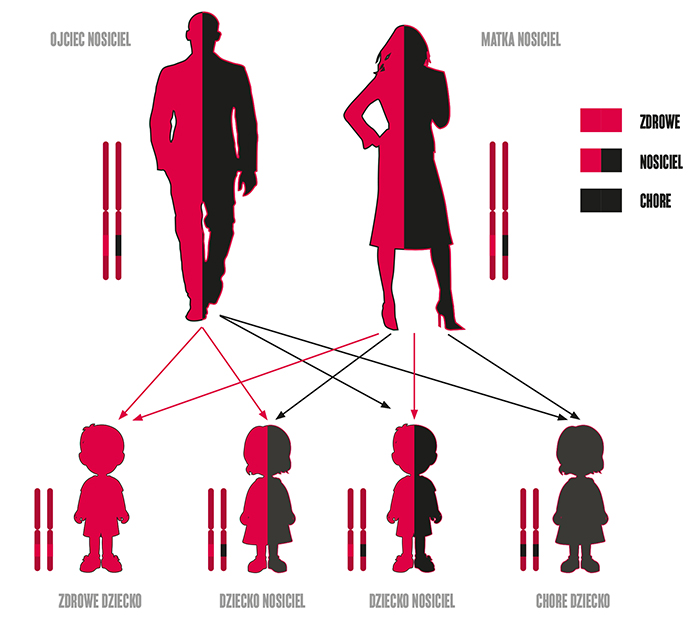
Chorzy skarżą się na niedowład ruchowy, zaburzenia połykania oraz niewydolność układu oddechowego. W krańcowym stadium choroby może u nich dość nawet do zgonu. Co ważne, rdzeniowy zanik mięśni nie powoduje u nich zaburzeń funkcji poznawczych mózgu i w podobnym stopniu do ich zdrowych rówieśników, odczuwają oni ból oraz odbierają bodźce dochodzące z otoczenia. SMA jest chorobą genetyczną, autosomalną, dziedziczoną w sposób recesywny. To znaczy, że aby się pojawiła konieczne jest odziedziczenie po obu rodzicach 2 kopii wadliwego genu — po jednej od każdego z nich. Jeżeli oboje rodziców jest nosicielami genu z mutacją, to prawdopodobieństwo urodzenia chorego dziecka wynosi 25 proc. Jeśli dziecko odziedziczy tylko jedną kopię wadliwego genu jest wprawdzie zdrowe, ale staje się nosicielem, który może przekazywać zmutowany gen swemu potomstwu.
Przyczyna SMA
Kluczem do wynalezienia na chorobę lekarstwa było odkrycie przyczyny jej rozwoju. Jak się okazało jest nią niedobór produkcji białka SMN (ang.: survival motor neuron) niezbędnego do prawidłowego funkcjonowania neuronów ruchowych. U ludzi informacja niezbędna do produkcji białka SMN zapisana jest w dwóch genach — SMN1 oraz SMN2 — umiejscowionych na chromosomie 5. Oba geny zawierają niemal identyczną informację w postaci sekwencji nukleotydowej, przy czym jedynie gen SMN1 koduje w pełni funkcjonalne białko SMN. Choroba SMA rozwija się w wyniku mutacji lub delecji obu kopii genu SMN1, prowadzących do zaprzestania produkcji funkcjonalnego białka SMN. W większości przypadków (ok 90–98%) następuje utrata (tzw. delecja) całych fragmentów kodujących. Jedynym źródłem białka SMN, w takim przypadku, pozostaje to wyprodukowane w oparciu o gen SMN2, jednak ilość ta nie zaspokaja rzeczywistego zapotrzebowania neuronów. W konsekwencji następuje obumieranie neuronów ruchowych prowadzące do atrofii mięśni.
Typy SMA
Wyróżniamy 5 postaci klinicznych SMA (SMA 0 — SMA IV). Rdzeniowy zanik mięśni typu 0 (SMA 0, prenatalny) to bardzo rzadka i skrajnie ciężka postać SMA. Objawy obecne są już w okresie życia płodowego, kiedy dziecko słabiej się rusza. Po urodzeniu jest wiotkie, obserwuje się u niego brak odruchów noworodkowych. Pojawiają się też zaburzenia ssania i połykania. Już w pierwszych dniach, a nawet godzinach życia rozwija się niewydolność oddechowa, która na ogół, już przed ukończeniem przez dziecko pierwszego miesiąca życia — prowadzi do śmierci. Najczęściej występującą, ostrą postacią niemowlęcą, manifestującą się już w pierwszych dniach lub miesiącach życia dziecka jest SMA typu I. W zależności od wieku, w którym pojawią się pierwsze objawy choroby wyodrębnia się postać SMA IA (początek przed 2 tygodniem życia), postać SMA IB (początek objawów między 2. tygodniem a 3. miesiącem życia) oraz postać SMA IC (objawy między 3. a 6. miesiącem życia). Diagnoza choroby może być opóźniona, ze względu na trudne do uchwycenia pierwsze objawy. Po kilku tygodniach lub miesiącach pozornego zdrowia, niepokój u rodziców i lekarzy wzbudza brak postępu w rozwoju ruchowym. Dziecko nie podnosi główki, nie przekręca się na boki, przestaje kopać nóżkami w kąpieli, łatwo męczy się przy jedzeniu i cicho płacze. Stwierdza się u niego wiotkość oraz niedowład czterokończynowy. Dzieci z SMA I często cierpią na znaczne deformacje kręgosłupa i klatki piersiowej. Pojawia się u nich rytmiczne drżenie palców, ciężka hipotonia, brak lub słaba kontrola ruchów głowy, niska masa mięśniowa, brak odruchów ścięgnistych, zespół opuszkowy, zaburzenia oddychania prowadzące do niewydolności oddechowej i już w przeciągu pierwszych 6 miesięcy życia konieczna jest u nich wentylacja mechaniczna. Do tego dochodzą zaburzania odkrztuszania, skłonność do nawracających infekcji dróg oddechowych, zaburzenia odżywienia. Jednym słowem — ich stan szybko się pogarsza. Dzieci z SMA I umierają najczęściej już przed ukończeniem 2. roku życia. Tylko niewielki odsetek z nich przeżyje kilka do kilkunastu lat. Z kolei SMA II jest pośrednią postacią choroby. Dzieci w pierwszym półroczu życia rozwijają się prawidłowo, potrafią samodzielnie siedzieć, jednak nigdy nie nabywają zdolności chodzenia. Pierwsze objawy choroby występują zwykle pomiędzy 18. miesiącem życia w postaci symetrycznego osłabienia mięśniowego. Pojawia się mowa nosowa i zaburzenia połykania. Z powodu ich wczesnego unieruchomienia już we wczesnych latach życia dochodzi do przykurczy stawowych, fascykulacji (drżenia pęczkowego języka), zniekształcenia klatki piersiowej i skoliozy. Chorzy mogą samodzielnie utrzymywać pozycję siedzącą, nie mogą natomiast stać ani chodzić. Ze względu na postęp choroby funkcje ruchowe pacjentów z SMA II są ograniczone do ruchów dłoni, przedramion i palców stóp. Szacuje się, że 75 proc. z nich dożywa 25. roku życia, przy czym główną przyczyną zgonów są powikłania oddechowe. W przypadku SMA III rozwój ruchowy dzieci w pierwszym roku życia przebiega prawidłowo. Potrafią samodzielnie siedzieć i chodzić. Dopiero pomiędzy 3. a 18. rokiem życia pojawia się u nich nieprawidłowy chód, fascykulacje, osłabione odruchy ścięgniste, trudności ze wstawaniem z pozycji leżącej i w chodzeniu po schodach. Zanik mięśni dotyczy przede wszystkim mięśni ksobnych, głównie obręczy biodrowej. W miarę postępu choroby trudności z poruszaniem się nasilają. Widoczny jest przerost rzekomy lub prawdziwy mięśni łydek i pośladków.
Wyróżniamy dwie podgrupy SMA III: pierwsza to SMA IIIA, w której objawy pojawiają się przed 3. rokiem życia. Dzieci już od początku źle chodzą, nie biegają, nie potrafią skakać, wstają z pomocą opierając się o przedmioty. Druga grupa obejmuje pacjentów z lepszym rozwojem ruchowym to SMA IIIB, a pierwsze objawy choroby występują po 3. roku życia. Rokowanie w SMA III co do przeżycia jest dobre. Chorzy z tą postacią rdzeniowego zaniku mięśni zwykle dożywają wieku dorosłego. Wielu jednak z nich wymaga, przynajmniej okresowo, nieinwazyjnego wsparcia oddechu. Najrzadszą postacią choroby jest SMA IV. Osłabienie siły mięśniowej pojawia się dopiero w wieku dorosłym, zwykle w trzeciej dekadzie życia. Początek objawów jest często trudny do wychwycenia. Pacjenci gorzej chodzą, często upadają. Dochodzi u nich do zaniku mięśni proksymalnych. Z czasem pojawiają się u nich trudności z wchodzeniem po schodach czy ze wstawaniem z pozycji kucznej. Przebieg choroby przebiega zwykle powoli, a chorzy mimo znacznego osłabienia ogólnej sprawności długo utrzymują zdolność samodzielnego chodzenia.
Dziedziczenie SMA

Przyczyna SMA
Kluczem do wynalezienia na chorobę lekarstwa było odkrycie przyczyny jej rozwoju. Jak się okazało jest nią niedobór produkcji białka SMN (ang.: survival motor neuron) niezbędnego do prawidłowego funkcjonowania neuronów ruchowych. U ludzi informacja niezbędna do produkcji białka SMN zapisana jest w dwóch genach — SMN1 oraz SMN2 — umiejscowionych na chromosomie 5. Oba geny zawierają niemal identyczną informację w postaci sekwencji nukleotydowej, przy czym jedynie gen SMN1 koduje w pełni funkcjonalne białko SMN. Choroba SMA rozwija się w wyniku mutacji lub delecji obu kopii genu SMN1, prowadzących do zaprzestania produkcji funkcjonalnego białka SMN. W większości przypadków (ok 90–98%) następuje utrata (tzw. delecja) całych fragmentów kodujących. Jedynym źródłem białka SMN, w takim przypadku, pozostaje to wyprodukowane w oparciu o gen SMN2, jednak ilość ta nie zaspokaja rzeczywistego zapotrzebowania neuronów. W konsekwencji następuje obumieranie neuronów ruchowych prowadzące do atrofii mięśni.
Typy SMA
Wyróżniamy 5 postaci klinicznych SMA (SMA 0 — SMA IV). Rdzeniowy zanik mięśni typu 0 (SMA 0, prenatalny) to bardzo rzadka i skrajnie ciężka postać SMA. Objawy obecne są już w okresie życia płodowego, kiedy dziecko słabiej się rusza. Po urodzeniu jest wiotkie, obserwuje się u niego brak odruchów noworodkowych. Pojawiają się też zaburzenia ssania i połykania. Już w pierwszych dniach, a nawet godzinach życia rozwija się niewydolność oddechowa, która na ogół, już przed ukończeniem przez dziecko pierwszego miesiąca życia — prowadzi do śmierci. Najczęściej występującą, ostrą postacią niemowlęcą, manifestującą się już w pierwszych dniach lub miesiącach życia dziecka jest SMA typu I. W zależności od wieku, w którym pojawią się pierwsze objawy choroby wyodrębnia się postać SMA IA (początek przed 2 tygodniem życia), postać SMA IB (początek objawów między 2. tygodniem a 3. miesiącem życia) oraz postać SMA IC (objawy między 3. a 6. miesiącem życia). Diagnoza choroby może być opóźniona, ze względu na trudne do uchwycenia pierwsze objawy. Po kilku tygodniach lub miesiącach pozornego zdrowia, niepokój u rodziców i lekarzy wzbudza brak postępu w rozwoju ruchowym. Dziecko nie podnosi główki, nie przekręca się na boki, przestaje kopać nóżkami w kąpieli, łatwo męczy się przy jedzeniu i cicho płacze. Stwierdza się u niego wiotkość oraz niedowład czterokończynowy. Dzieci z SMA I często cierpią na znaczne deformacje kręgosłupa i klatki piersiowej. Pojawia się u nich rytmiczne drżenie palców, ciężka hipotonia, brak lub słaba kontrola ruchów głowy, niska masa mięśniowa, brak odruchów ścięgnistych, zespół opuszkowy, zaburzenia oddychania prowadzące do niewydolności oddechowej i już w przeciągu pierwszych 6 miesięcy życia konieczna jest u nich wentylacja mechaniczna. Do tego dochodzą zaburzania odkrztuszania, skłonność do nawracających infekcji dróg oddechowych, zaburzenia odżywienia. Jednym słowem — ich stan szybko się pogarsza. Dzieci z SMA I umierają najczęściej już przed ukończeniem 2. roku życia. Tylko niewielki odsetek z nich przeżyje kilka do kilkunastu lat. Z kolei SMA II jest pośrednią postacią choroby. Dzieci w pierwszym półroczu życia rozwijają się prawidłowo, potrafią samodzielnie siedzieć, jednak nigdy nie nabywają zdolności chodzenia. Pierwsze objawy choroby występują zwykle pomiędzy 18. miesiącem życia w postaci symetrycznego osłabienia mięśniowego. Pojawia się mowa nosowa i zaburzenia połykania. Z powodu ich wczesnego unieruchomienia już we wczesnych latach życia dochodzi do przykurczy stawowych, fascykulacji (drżenia pęczkowego języka), zniekształcenia klatki piersiowej i skoliozy. Chorzy mogą samodzielnie utrzymywać pozycję siedzącą, nie mogą natomiast stać ani chodzić. Ze względu na postęp choroby funkcje ruchowe pacjentów z SMA II są ograniczone do ruchów dłoni, przedramion i palców stóp. Szacuje się, że 75 proc. z nich dożywa 25. roku życia, przy czym główną przyczyną zgonów są powikłania oddechowe. W przypadku SMA III rozwój ruchowy dzieci w pierwszym roku życia przebiega prawidłowo. Potrafią samodzielnie siedzieć i chodzić. Dopiero pomiędzy 3. a 18. rokiem życia pojawia się u nich nieprawidłowy chód, fascykulacje, osłabione odruchy ścięgniste, trudności ze wstawaniem z pozycji leżącej i w chodzeniu po schodach. Zanik mięśni dotyczy przede wszystkim mięśni ksobnych, głównie obręczy biodrowej. W miarę postępu choroby trudności z poruszaniem się nasilają. Widoczny jest przerost rzekomy lub prawdziwy mięśni łydek i pośladków.
Wyróżniamy dwie podgrupy SMA III: pierwsza to SMA IIIA, w której objawy pojawiają się przed 3. rokiem życia. Dzieci już od początku źle chodzą, nie biegają, nie potrafią skakać, wstają z pomocą opierając się o przedmioty. Druga grupa obejmuje pacjentów z lepszym rozwojem ruchowym to SMA IIIB, a pierwsze objawy choroby występują po 3. roku życia. Rokowanie w SMA III co do przeżycia jest dobre. Chorzy z tą postacią rdzeniowego zaniku mięśni zwykle dożywają wieku dorosłego. Wielu jednak z nich wymaga, przynajmniej okresowo, nieinwazyjnego wsparcia oddechu. Najrzadszą postacią choroby jest SMA IV. Osłabienie siły mięśniowej pojawia się dopiero w wieku dorosłym, zwykle w trzeciej dekadzie życia. Początek objawów jest często trudny do wychwycenia. Pacjenci gorzej chodzą, często upadają. Dochodzi u nich do zaniku mięśni proksymalnych. Z czasem pojawiają się u nich trudności z wchodzeniem po schodach czy ze wstawaniem z pozycji kucznej. Przebieg choroby przebiega zwykle powoli, a chorzy mimo znacznego osłabienia ogólnej sprawności długo utrzymują zdolność samodzielnego chodzenia.
Dziedziczenie SMA