Kiedy należy myśleć o niedoborze kwaśnej lipazy lizosomalnej

Kwaśna lipaza lizosomalna (lysosomal acid lipase – LAL) zwana jest również kwaśną esterazą lub hydrolazą estrów cholesterolu. Jej niedobór kwaśnej lipazy lizosomalnej (LAL deficiency – LAL-D) jest bardzo rzadko rozpoznawanym schorzeniem i dlatego prawie nieznanym.
W zależności od pochodzenia etnicznego i strefy geograficznej częstość jego występowania oszacowano na od 1/40 000 do 1/300 000 osób. Według badań niemieckich częstość występowania LAL-D wynosi 1/40 000 osób. Jak wynika z tych szacunków, choroba może być niejednokrotnie niezdiagnozowana.
Jaka jest rola LAL
– Enzym LAL katalizuje przebieg reakcji odszczepiania kwasów tłuszczowych z trójglicerydów i estrów cholesterolu. Znajduje się w lizosomach, stąd nazwa kwaśna lipaza lizosomalna – wyjaśnia prof. dr hab. n. med. Anna Tylki-Szymańska z Kliniki Pediatrii, Żywienia i Chorób Metabolicznych Instytutu „Pomnik – Centrum Zdrowia Dziecka”. Uwolnione kwasy tłuszczowe są wiązane przez białko transportujące (FAB) i kierowane do rożnych organelli komórkowych. Wolny cholesterol jest natomiast przenoszony do błony komórkowej lub siatki endoplazmatycznej, gdzie hamuje syntezę cholesterolu całkowitego i frakcji LDL oraz stymuluje syntezę estrów cholesterolu. Białko LAL jest kodowane przez gen LIPA zlokalizowany na długim ramieniu chromosomu 10 w pozycji 10q23.31. Mutacje w tym genie powodują niedobór aktywności LAL prowadzący do odkładania estrów cholesterolu i trójglicerydów m.in. w komórkach wątrobowych, śledzionie oraz makrofagach. Najczęstszą mutacją jest c.894G>A, stwierdzana u ponad 50 proc. chorych na LAL-D.
– Niedobór LAL sprawia, że w lizosomach odkładają się estry cholesterolowe i trójglicerydy. Zjawisko to jest widoczne głównie w hepatocytach, ale zachodzi również w makrofagach. Dochodzi wtedy do zaburzenia funkcji lizosomów, które tracą zdolność regulacji metabolizmu lipidowego i wysyłają na zewnątrz do organelli komórkowych fałszywą informację o rzekomym niedoborze cholesterolu. Niedobór LAL to groźna choroba – wieloukładowa, przewlekła i postępująca. Jej rozpoznanie jest bardzo ważne, ponieważ dostępne jest specyficzne leczenie – tłumaczy prof. dr hab. n. med. Marek Hartleb, kierownik Katedry i Kliniki Gastroenterologii i Hepatologii Śląskiego Uniwersytetu Medycznego w Katowicach.
– Niedobór LAL, jak większość chorób metabolicznych, jest chorobą monogenową, dziedziczy się ją autosomalnie recesywnie. Poznano 47 mutacji powodujących deficyt LAL. Zachodzi związek między ciężkością kliniczną LAL-D a typem mutacji determinującej aktywność LAL – mówi prof. Anna Tylki-Szymańska.
Choroba wieloukładowa
W zależności od stopnia niedoboru LAL wyróżnia się dwa fenotypy: postać wczesną, zwaną chorobą Wolmana, występującą w okresie niemowlęcym, o bardzo ciężkim przebiegu, charakteryzującą się brakiem lub śladową aktywnością LAL, oraz postać późną – chorobę spichrzania estrów cholesterolu (cholesteryl ester storage disease – CESD), zwykle występującą u starszych dzieci, lecz również u dorosłych. Badania wskazują, że 15–17 proc. pacjentów z rozpoznaniem tej choroby ma powyżej 15 lat. Postęp choroby jest szybki – od pojawienia się pierwszych objawów do zaawansowanego włóknienia bądź marskości wątroby mija zaledwie kilka lat. Niedobór LAL jest stosunkowo niedawno poznaną chorobą. W 1956 r. neuropatolog Wolman opisał przypadek niemowlęcia z hepatosplenomegalią i zwapnieniem nadnerczy. Z kolei w 1963 r. Fredrickson zaprezentował przypadek LAL-D u dorosłego.
– Objawy kliniczne w chorobie Wolmana pojawiają się w pierwszych tygodniach życia. Są to uporczywe wymioty, hepatosplenomegalia, wzdęcia jelit i niedokrwistość. Przebieg kliniczny jest dramatyczny, a zgon pacjenta, wobec którego jesteśmy kompletnie bezradni, następuje zwykle w pierwszym półroczu życia. W obrazie klinicznym uwagę zwracają zwapnienia powiększonych nadnerczy, podwyższenie aktywności transaminaz, niedokrwistość i zwiększone stężenie trójglicerydów – mówi prof. Anna Tylki-Szymańska. Choroba spichrzania estrów cholesterolu przebiega dużo łagodniej niż choroba Wolmana.
Obserwuje się mierne powiększenie śledziony i wątroby, uchwytne w pierwszych latach życia lub dopiero u dorosłych. Znane są też postacie bezobjawowe, ponieważ pacjent poza dyskomfortem wynikającym z powiększenia wątroby może nie odczuwać innych dolegliwości. Wykładniki laboratoryjne to podwyższone stężenie cholesterolu całkowitego oraz LDL, obniżone stężenie frakcji HDL cholesterolu oraz podwyższona aktywność transaminaz. Istnieje jeszcze jeden biomarker tej choroby – chitotriozydaza. Niedobór LAL jest chorobą wieloukładową. Z powodu zwiększonego stężenia cholesterolu LDL i zmniejszonego stężenia HDL pojawia się miażdżyca w młodym wieku z wszystkimi powikłaniami sercowo- naczyniowymi. Podwyższone stężenie cholesterolu nie reaguje na leczenie statynami.
– Wątroba jest narządem często zajmowanym – w ponad 80 proc. przypadków. Stłuszczenie wątroby i zwiększone stężenie cholesterolu sprawiają, że LAL-D naśladuje niealkoholową chorobę stłuszczeniową wątroby (non-alcoholic fatty liver disease – NAFLD) w przebiegu zespołu metabolicznego. W badaniu histologicznym wątroby zwraca jednak uwagę duży udział stłuszczenia drobnokropelkowego. Ponadto LAL-D odpowiada za zwiększoną aktywność aminotransferazy alaninowej, która bywa często wyższa niż w NAFLD – mówi prof. Marek Hartleb.
Rozpoznanie LAL-D czasami jest ustalane w stadium marskości wątroby u młodego człowieka. U niektórych pacjentów problemem jest olbrzymia śledziona z powodu odkładania estrów cholesterolowych w makrofagach śledzionowych. U chorych istnieje skłonność do samoistnych pęknięć śledziony i głębokiej trombocytopenii.
Jak diagnozować LAL-D
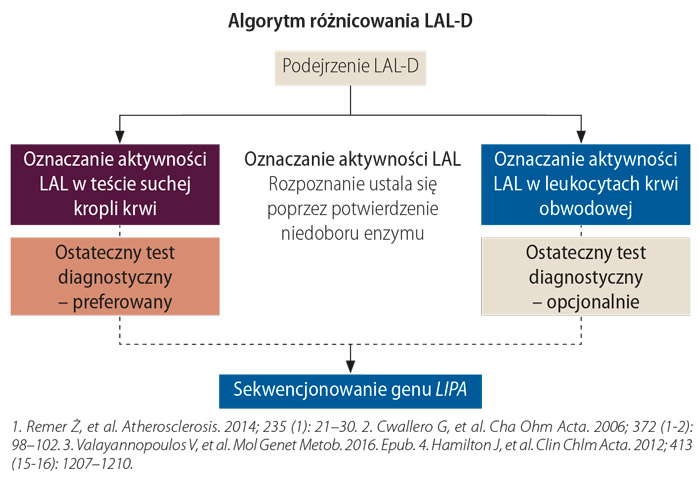
– Jeżeli pacjent ma podwyższone transaminazy, dyslipidemię, powiększone wątrobę i śledzionę, jesteśmy upoważnieni do wykonania badań w kierunku LAL-D w celu wykluczenia choroby, a nie koniecznie potwierdzenia podejrzenia. LAL-D występuje częściej, niż ją rozpoznajemy czy w ogóle o niej myślimy. Jeśli choroba nie prowadzi do dużego dyskomfortu u pacjenta, to taka sytuacja nie motywuje lekarza do poszukiwania patologii – uważa prof. Anna Tylki-Szymańska. Można zbadać aktywność LAL w leukocytach krwi czy hodowanych fibroblastach skory. Coraz częściej odstępuje się od tej metody, choć jest ona bardzo wiarygodna i czuła. Ostatnio opracowano metodę z wykorzystaniem suchej kropli krwi. Diagnostyka na podstawie materiału z suchej kropli krwi pozwala na szybkie wykluczenie lub potwierdzenie LAL-D. Istnieje ryzyko wyników fałszywie dodatnich i dlatego konieczna jest analiza molekularna.
– Generalnie wszystkie choroby metaboliczne, które wynikają z deficytów aktywności enzymu czy deficytów funkcji innych białek, zawsze należy starać się potwierdzić badaniem molekularnym – podkreśla prof. Anna Tylki-Szymańska.
Deficyt kwaśnej esterazy można również potwierdzić poprzez wykazanie spichrzenia estrów cholesterolu, trójglicerydów i cholesterolu za pomocą chromatografii cienkowarstwowej. Obecność igieł wolnego cholesterolu w bioptacie wątroby mocno sugeruje LAL-D.
Leczenie przyczynowe – tylko enzymatyczna terapia zastępcza
Stosowanie preparatów hamujących syntezę cholesterolu jest postępowaniem wyłącznie objawowym i nie zapobiega procesowi marskiemu w wątrobie.
– Leczenie przyczynowe to enzymatyczna terapia substytucyjna. Podejmowano próby przeszczepu szpiku u pacjentów z chorobą Wolmana, ale jest to tak ciężka choroba, że nie udaje się doprowadzić do bezpiecznego wykonania procedury – mówi prof. Anna Tylki-Szymańska.
Enzymatyczna terapia zastępcza enzymem sebelipazą-a została zatwierdzona w Stanach Zjednoczonych i Unii Europejskiej w 2015 r. Podaje się ją raz w tygodniu we wlewie dożylnym pacjentom z szybko postępującą chorobą w pierwszych 6 miesiącach życia. U osób z mniej agresywnym wariantem LAL-D lek podaje się w odstępach dwutygodniowych. Leczenie enzymatyczne daje dobre efekty, prowadzi do normalizacji aktywności transaminaz i przywrócenia homeostazy lipidów. Ta terapia nie jest jeszcze w Polsce refundowana.
Tekst opublikowano w „Kurierze Medycznym” 1/2021. Czasopismo można zamówić na stronie: www.termedia.pl/km/prenumerata.
Przeczytaj także: „Jak leczyć zaburzenia lipidowe w 2021 r. – nowe wytyczne Polskiego Towarzystwa Lipidologicznego”.

Jaka jest rola LAL
– Enzym LAL katalizuje przebieg reakcji odszczepiania kwasów tłuszczowych z trójglicerydów i estrów cholesterolu. Znajduje się w lizosomach, stąd nazwa kwaśna lipaza lizosomalna – wyjaśnia prof. dr hab. n. med. Anna Tylki-Szymańska z Kliniki Pediatrii, Żywienia i Chorób Metabolicznych Instytutu „Pomnik – Centrum Zdrowia Dziecka”. Uwolnione kwasy tłuszczowe są wiązane przez białko transportujące (FAB) i kierowane do rożnych organelli komórkowych. Wolny cholesterol jest natomiast przenoszony do błony komórkowej lub siatki endoplazmatycznej, gdzie hamuje syntezę cholesterolu całkowitego i frakcji LDL oraz stymuluje syntezę estrów cholesterolu. Białko LAL jest kodowane przez gen LIPA zlokalizowany na długim ramieniu chromosomu 10 w pozycji 10q23.31. Mutacje w tym genie powodują niedobór aktywności LAL prowadzący do odkładania estrów cholesterolu i trójglicerydów m.in. w komórkach wątrobowych, śledzionie oraz makrofagach. Najczęstszą mutacją jest c.894G>A, stwierdzana u ponad 50 proc. chorych na LAL-D.
– Niedobór LAL sprawia, że w lizosomach odkładają się estry cholesterolowe i trójglicerydy. Zjawisko to jest widoczne głównie w hepatocytach, ale zachodzi również w makrofagach. Dochodzi wtedy do zaburzenia funkcji lizosomów, które tracą zdolność regulacji metabolizmu lipidowego i wysyłają na zewnątrz do organelli komórkowych fałszywą informację o rzekomym niedoborze cholesterolu. Niedobór LAL to groźna choroba – wieloukładowa, przewlekła i postępująca. Jej rozpoznanie jest bardzo ważne, ponieważ dostępne jest specyficzne leczenie – tłumaczy prof. dr hab. n. med. Marek Hartleb, kierownik Katedry i Kliniki Gastroenterologii i Hepatologii Śląskiego Uniwersytetu Medycznego w Katowicach.
– Niedobór LAL, jak większość chorób metabolicznych, jest chorobą monogenową, dziedziczy się ją autosomalnie recesywnie. Poznano 47 mutacji powodujących deficyt LAL. Zachodzi związek między ciężkością kliniczną LAL-D a typem mutacji determinującej aktywność LAL – mówi prof. Anna Tylki-Szymańska.
Choroba wieloukładowa
W zależności od stopnia niedoboru LAL wyróżnia się dwa fenotypy: postać wczesną, zwaną chorobą Wolmana, występującą w okresie niemowlęcym, o bardzo ciężkim przebiegu, charakteryzującą się brakiem lub śladową aktywnością LAL, oraz postać późną – chorobę spichrzania estrów cholesterolu (cholesteryl ester storage disease – CESD), zwykle występującą u starszych dzieci, lecz również u dorosłych. Badania wskazują, że 15–17 proc. pacjentów z rozpoznaniem tej choroby ma powyżej 15 lat. Postęp choroby jest szybki – od pojawienia się pierwszych objawów do zaawansowanego włóknienia bądź marskości wątroby mija zaledwie kilka lat. Niedobór LAL jest stosunkowo niedawno poznaną chorobą. W 1956 r. neuropatolog Wolman opisał przypadek niemowlęcia z hepatosplenomegalią i zwapnieniem nadnerczy. Z kolei w 1963 r. Fredrickson zaprezentował przypadek LAL-D u dorosłego.
– Objawy kliniczne w chorobie Wolmana pojawiają się w pierwszych tygodniach życia. Są to uporczywe wymioty, hepatosplenomegalia, wzdęcia jelit i niedokrwistość. Przebieg kliniczny jest dramatyczny, a zgon pacjenta, wobec którego jesteśmy kompletnie bezradni, następuje zwykle w pierwszym półroczu życia. W obrazie klinicznym uwagę zwracają zwapnienia powiększonych nadnerczy, podwyższenie aktywności transaminaz, niedokrwistość i zwiększone stężenie trójglicerydów – mówi prof. Anna Tylki-Szymańska. Choroba spichrzania estrów cholesterolu przebiega dużo łagodniej niż choroba Wolmana.
Obserwuje się mierne powiększenie śledziony i wątroby, uchwytne w pierwszych latach życia lub dopiero u dorosłych. Znane są też postacie bezobjawowe, ponieważ pacjent poza dyskomfortem wynikającym z powiększenia wątroby może nie odczuwać innych dolegliwości. Wykładniki laboratoryjne to podwyższone stężenie cholesterolu całkowitego oraz LDL, obniżone stężenie frakcji HDL cholesterolu oraz podwyższona aktywność transaminaz. Istnieje jeszcze jeden biomarker tej choroby – chitotriozydaza. Niedobór LAL jest chorobą wieloukładową. Z powodu zwiększonego stężenia cholesterolu LDL i zmniejszonego stężenia HDL pojawia się miażdżyca w młodym wieku z wszystkimi powikłaniami sercowo- naczyniowymi. Podwyższone stężenie cholesterolu nie reaguje na leczenie statynami.
– Wątroba jest narządem często zajmowanym – w ponad 80 proc. przypadków. Stłuszczenie wątroby i zwiększone stężenie cholesterolu sprawiają, że LAL-D naśladuje niealkoholową chorobę stłuszczeniową wątroby (non-alcoholic fatty liver disease – NAFLD) w przebiegu zespołu metabolicznego. W badaniu histologicznym wątroby zwraca jednak uwagę duży udział stłuszczenia drobnokropelkowego. Ponadto LAL-D odpowiada za zwiększoną aktywność aminotransferazy alaninowej, która bywa często wyższa niż w NAFLD – mówi prof. Marek Hartleb.
Rozpoznanie LAL-D czasami jest ustalane w stadium marskości wątroby u młodego człowieka. U niektórych pacjentów problemem jest olbrzymia śledziona z powodu odkładania estrów cholesterolowych w makrofagach śledzionowych. U chorych istnieje skłonność do samoistnych pęknięć śledziony i głębokiej trombocytopenii.
Jak diagnozować LAL-D
– Jeżeli pacjent ma podwyższone transaminazy, dyslipidemię, powiększone wątrobę i śledzionę, jesteśmy upoważnieni do wykonania badań w kierunku LAL-D w celu wykluczenia choroby, a nie koniecznie potwierdzenia podejrzenia. LAL-D występuje częściej, niż ją rozpoznajemy czy w ogóle o niej myślimy. Jeśli choroba nie prowadzi do dużego dyskomfortu u pacjenta, to taka sytuacja nie motywuje lekarza do poszukiwania patologii – uważa prof. Anna Tylki-Szymańska. Można zbadać aktywność LAL w leukocytach krwi czy hodowanych fibroblastach skory. Coraz częściej odstępuje się od tej metody, choć jest ona bardzo wiarygodna i czuła. Ostatnio opracowano metodę z wykorzystaniem suchej kropli krwi. Diagnostyka na podstawie materiału z suchej kropli krwi pozwala na szybkie wykluczenie lub potwierdzenie LAL-D. Istnieje ryzyko wyników fałszywie dodatnich i dlatego konieczna jest analiza molekularna.
– Generalnie wszystkie choroby metaboliczne, które wynikają z deficytów aktywności enzymu czy deficytów funkcji innych białek, zawsze należy starać się potwierdzić badaniem molekularnym – podkreśla prof. Anna Tylki-Szymańska.
Deficyt kwaśnej esterazy można również potwierdzić poprzez wykazanie spichrzenia estrów cholesterolu, trójglicerydów i cholesterolu za pomocą chromatografii cienkowarstwowej. Obecność igieł wolnego cholesterolu w bioptacie wątroby mocno sugeruje LAL-D.
Leczenie przyczynowe – tylko enzymatyczna terapia zastępcza
Stosowanie preparatów hamujących syntezę cholesterolu jest postępowaniem wyłącznie objawowym i nie zapobiega procesowi marskiemu w wątrobie.
– Leczenie przyczynowe to enzymatyczna terapia substytucyjna. Podejmowano próby przeszczepu szpiku u pacjentów z chorobą Wolmana, ale jest to tak ciężka choroba, że nie udaje się doprowadzić do bezpiecznego wykonania procedury – mówi prof. Anna Tylki-Szymańska.
Enzymatyczna terapia zastępcza enzymem sebelipazą-a została zatwierdzona w Stanach Zjednoczonych i Unii Europejskiej w 2015 r. Podaje się ją raz w tygodniu we wlewie dożylnym pacjentom z szybko postępującą chorobą w pierwszych 6 miesiącach życia. U osób z mniej agresywnym wariantem LAL-D lek podaje się w odstępach dwutygodniowych. Leczenie enzymatyczne daje dobre efekty, prowadzi do normalizacji aktywności transaminaz i przywrócenia homeostazy lipidów. Ta terapia nie jest jeszcze w Polsce refundowana.
Tekst opublikowano w „Kurierze Medycznym” 1/2021. Czasopismo można zamówić na stronie: www.termedia.pl/km/prenumerata.
Przeczytaj także: „Jak leczyć zaburzenia lipidowe w 2021 r. – nowe wytyczne Polskiego Towarzystwa Lipidologicznego”.