
Introduction
Inflammatory bowel disease (IBD) is a heterogenous group of chronic inflammations in the gastrointestinal tract, which traditionally consists of two types: Crohn’s disease (CD) and ulcerative colitis (UC). They differ when it comes to clinical, endoscopic, and histopathological changes. The aetiology of IBD is not fully known, but it has been linked to genetic, immunological, and environmental factors playing a role in the development of the disease. Interaction between genetic and environmental factors leads to immunological response, which has an impact on microbiota and causes inflammation of the intestine [1–3]. Proinflammatory cytokines mediate the relation between lymphoid, inflammatory, and haematopoietic cells. Activated intraepithelial lymphocytes (IELs) within intestines are the first-line defence. They produce interleukin 15 (IL-15), which stimulates macrophages, neutrophils, eosinophils, and CD8+ T lymphocytes in the lamina propria [4]. As a result, proinflammatory cytokines such as IL-1β, IL-2, IL-6, IL-8 , and IL-12 and tumor necrosis factor α (TNF-α) appear. TNF-α inhibits anti-inflammatory transforming growth factor β (TGF-β), which induces fibrotic processes. Gut dysbiosis, including the role of bacteria, fungi, viruses, and other microorganisms, has also been studied extensively, proving a decrease in biodiversity, known as α diversity [5]. Gevers et al. [6] suggested that increased Veillonellaceae, Pasteurellacaea, Enterobacteriaceae, and Fusobacteriaceae and decreased Bacteroidales, Erysipelotrichales, and Clostridiales, strongly correlate with disease status.
On the other hand, cell wall components of the fungi can also trigger the innate immune response through receptors such as dectin-1 (a C-type lectin receptor), Toll-like receptors (TLR2 and TLR4), components of the complement system, and members of the scavenger receptor family (CD5, SCARF1, and CD36) [7]. As a result, different molecules (CARD9, IL-17, IL-22, ITAM, NFAT, and NF-kB) are ejected. When it comes to the role of viruses, the data is not straightforward. It seems that different viral strains together with gene mutations (ATG16L1/an autophagy gene, TLR3 and TLR7) may have both protective and aggravating influence on intestinal inflammation. Nonetheless, recently most studies have been focused on the genetic impact on IBD pathogenesis. More than 200 single nucleotide polymorphisms (SNPs) have already been described. Based on alterations in different molecules, new subtypes of IBD have also been proposed.
The aim of this article is to present cellular mechanisms leading to IBD with a focus on new forms of this disorder.
The basic diagram showing the impact of different pathogenetic factors and their most important molecular pathways in patients with IBD is presented in Figure 1.
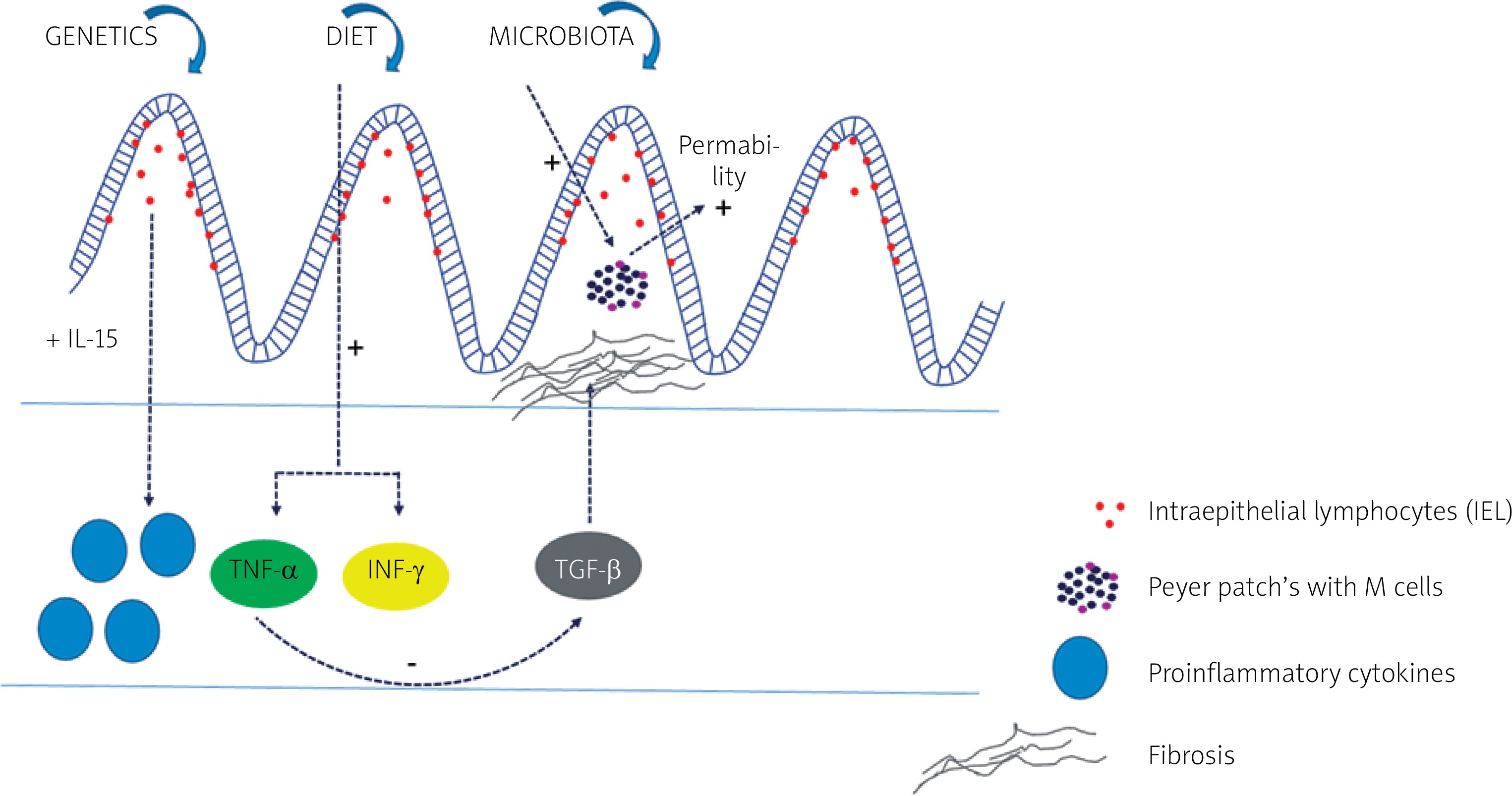
Crohn’s disease and ulcerative colitis – genetic and molecular background
The first SNP to be associated with IBD was discovered in 2001 by genome-wide association studies (GWAS). It was nucleotide-binding oligomerisation domain (NOD2)/caspase-activating recruitment domain (CARD4), which belongs to the pattern recognition receptor (PRR) group of molecules. In 2010, a meta-analysis was performed, which identified 71 susceptibility loci in the pathogenesis of CD [8]. For UC a meta-analysis was conducted in 2011 detecting 47 susceptibility loci [9]. Nowadays, more than 200 SNPs have been found and described [10, 11]. They belong to four different groups: PRR, genes associated with mucosal membrane integrity, autophagy genes, and genes for pro- and anti-inflammatory cytokines. The working mechanism of NOD2/CARD15 protein is related to activation of pathways of NF-κB and MAP kinase, which release proinflammatory cytokines. Genes that code transporters for carnitine and cations have also been discovered as IBD SNPs [12]. They are located within the IBD5 gene, which codes two transporters for carnitine 2: OCTN1 and OCTN2. The first one decreases transport of carnitine and increases transport of cations, while the second one inhibits carnitine uptake [13]. DLG5 (discs large homolog 5) gene, which belongs to MAGUK (membrane-associated guanylate kinase), is another SNP associated with mucosal membrane integrity. The protein coded by this gene has an impact on adhesion molecules and cytoskeleton-influencing transmembrane signalling, as well as maintaining a proper structure of intestinal epithelium. DLG5 (R30Q) is responsible for the coexistence of CD and UC [14]. Autophagy genes are very important because this process plays a crucial role in apoptosis. In 2007, Hampe et al. [15] proved that polymorphism in Thr300Ala in the ATG16L1 gene (bacterial autophagy) is associated with CD. A meta-analysis has been conducted revealing that variant rs2241880/T300A of this gene may contribute to CD development in childhood; however, it has not been confirmed for UC [13]. Therefore, it seems that ATG16L1 is an SNP gene for CD exclusively. When it comes to genes for pro- and anti-inflammatory cytokines, the best known and confirmed mutation is the one in IL-10 and its receptors predisposing to IBD manifestation in early childhood.
It is also important to mention the impact of diet in the pathogenesis of CD and UC. Although most of the data comes from mouse trials and their extrapolation to humans is difficult, findings are very promising and interesting particularly in regard to therapeutic methods. Many researches have also proven that a “western diet” characterised by high fat, sugar, gluten, dairy, and food additive intake accompanied by low fibre exposure may contribute to IBD [16, 17]. These alimentary factors may have a synergistic negative impact on intestinal microbiota and the intestinal barrier. Consequently, dysbiosis and increased permeability of the intestinal barrier induce abnormal immune response and inflammation [18]. For example, a high-fat and high-sugar diet decreases butyrate production and expression of its receptor (GPR43) [19]. Butyrate plays a protective role on the mucosal layer down-regulating inflammation by suppressing transcription of cytokines and increasing differentiation of lamina propria [20]. It also upregulates defensins and cathelicidin peptides with an antibacterial effect [21]. Martinez-Medina et al. [19] published data suggesting that a high-fat and high-sugar diet increased susceptibility to adherent invasive E. coli (AIEC) strains in IBD patients. In normal conditions AIEC cannot induce inflammation by itself. The pathogenicity appears only if ligand carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM 6), acting as a host cell receptor for AIEC adhesion, is presented by intestinal epithelial and M cells. It seems that high intake of fat and sugar may promote colonisation in genetically predisposed patients, expressing a basal level of CEACAM6. The negative effect of a HF/HS diet intensified by low dietary fibre intake leads to increased permeability of the mucous layer due to its catabolism. This process results in extensive contact between epithelium and pathogenic luminal bacteria [19]. Gluten may also induce ileitis, leading to intestinal permeability, which has been identified as a factor associated with relapse in CD [21]. Wheat contains α-amylase and trypsin inhibitors with the potential to activate human dendritic cells and macrophages driving intestinal inflammation [22].
We describe above only examples of the possible negative impact of diet on the pathogenesis of IBD. These mechanisms, however, are complex and multifactorial, but their detailed discussion is not the subject of this article.
Very early onset IBD and infantile IBD
Initially, due to the unique disease characteristics, the Paris Modification of the Montreal Classification system distinguished only one subgroup of paediatric IBD patients at age below 10 years [1]. More recently, according to the phenotypes as well as the presence of monogenic defects, a more detailed classification has been suggested. Subgroups of paediatric-onset IBD (< 17 years old), early onset IBD (< 10 years old), very-early onset IBD (VEO-IBD, < 6 years old), infantile onset IBD (< 2 years old), and neonatal onset IBD (< 28 days old) have been proposed [2–4]. Presentations of IBD at a very young age are quite different from those in adolescents and adults. In most cases, the disease is initially restricted to the colon, resistant to immunosuppressive or surgical treatment, and frequently associated with additional immunodeficiency disorders. The majority of patients with early IBD present with positive family history and significantly higher rates of affected first-degree relatives than others. All these reasons make researchers consider genetic susceptibility and environmental components as important factors in VEO-IBD [4, 5]. For example, single genetic mutations in the interleukin-10 (IL-10) or the IL-10 receptor (IL-10RA), impairing its immunological signalling pathway, have been found to be associated with these early IBD entities [6, 7].
Very early onset IBD caused by monogenic disorders, especially the ones associated with IL-10 RA deficiency, results in extraordinarily severe forms of IBD [8–10]. Patients with IL-10RA deficiency present with very severe gastrointestinal symptoms, such as acute colitis, severe perianal fistulae, and perirectal abscesses and recurrent clinical sepsis. In many cases monogenic IBD is accompanied with folliculitis and/or arthritis. The first symptoms may even appear in the first weeks of life [8–13]. This IL-10RA-deficient VEO-IBD is also known to be refractory to conventional medical management and may require very specific therapies such as bone marrow or stem cell transplantation [4, 5, 13]. Phenotypic characterisation of VEO-IBD, due to mutations in IL10 and IL10RA as well as possible treatment, have been extensively studied and described by the Genius Working Group, who collected the data of infants with a proven defect of the IL10 axis [14]. VEO-IBD constitutes 3–15% of paediatric IBD patients, while monogenic IL-10RA-deficient VEO-IBD is estimated at 10% of all VEO-IBD patients [15].
IL-10 is critical in maintaining the balance of the immune system through restrictions of the secretion of pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-12, and it controls both the differentiation and proliferation of macrophages and T and B cells. This is especially important within the intestine because there are over a hundred trillion bacteria constantly stimulating the immune system. Without IL-10 suppression, patients develop very severe intestinal inflammation [4, 5].
The genetic aetiology of monogenic early IBD is not only restricted to the IL-10 pathway. Large-scale genome-wide association studies (GWAS) have detected over 230 risk loci associated with VEO-IBD; however, the contribution of the majority of them in IBD manifestations remains unclear [16]. Whole exome sequencing (WES) has greatly expanded the list of genes associated with IBD risk, and currently almost 60 monogenic mutations related to VEO-IBD and IBD-like colitis have been identified. Among many others, mutations in NCF2, XIAP, LRBA, or TTC7 can be indicated. For example, mutations in the gene-encoding X-linked inhibitor of apoptosis (XIAP) in infants, like those engaged in IL-10, are connected with very severe IBD course. Recently, Zheng et al. have published three cases of infantile-onset IBD patients who had mutations in tumour necrosis factor α-induced protein 3 (TNFAIP3). It may be a new molecular pathway associated with infantile-onset IBD.
In conclusion, all studies and observations seem to indicate that monogenic VEO-IBD is a distinct, unique disease entity characterised by very aggressive course, and high morbidity and mortality, which requires a specific treatment strategy. Well-designed further studies are needed to confirm this theory.
IBD-PSC
It has been estimated that 75% of children with PSC also have IBD [23], while the presence of PSC in UC patients is approximately 2.4–7.5%, and in CD it is about 3.4%. The phenotype of IBD-PSC is unique and is characterised by active inflammation with poor clinical symptoms. Rectal sparing as well as backwash ileitis are also common. Patients have higher risk of developing colorectal cancer even if they are in remission. Based on these observations, coexistence of IBD and PSC has been considered as a distinct entity (different form of IBD). GWAS have even identified common genetic regions specific for this disorder. It is believed that the major histocompatibility complex (MHC) is the main genetic factor associated with PSC-UC. In 2007 a Scandinavian group compared PSC-UC and UC patients, revealing that distinct HLA class II was associated with the PSC-UC group. In 2011 Janse et al. [24] identified another three genetic links involving IL-2, REL, and CARD9 pathways as UC susceptibility loci associated with PSC. Further molecular changes that may predispose to PSC-IBD include SNPs and post-transcriptional gene regulators such as microRNA (miRNA), as well as different cytokines and a variety of autoantibodies. TNF-α and pANCA (PR3-ANCA) are also highly suspected to play a key role in the development of this disorder, especially because both PSC and IBD are strongly associated with autoimmune dysfunctions. This subject is currently being studied and no statements have been established so far.
Conclusions
IBD is a heterogenous disorder. It has been extensively studied for years to better understand its pathogenesis and to introduce novel therapies. Based on recent genomic researches and clinical observations, new phenotypes and molecular pathways have been discovered. However, it is still believed that IBD includes two distinct types: CD and UC, but their genetic background seems to be more compound than was previously thought. Perhaps, in the future new forms of IBD will be established. Nonetheless, even if considered as one disorder, it is evident that IBD has various molecular faces.










