
Introduction
Congenital adrenal hyperplasia (CAH) is an autosomal recessive genetic disorder. Majority of the cases are due to mutations in the CY21A2 gene [1]. The incidence of CAH is reported to be 1 in 2036 (Chennai), 1 in 2600 (Andhra), 1 in 2800 (Bengaluru), and 1 in 9983 (Mumbai) [2–4]. There are standard guidelines from the Endocrine Society that should be adopted by clinicians caring for children with CAH [1, 5]. Clinicians caring for children with CAH face a multitude of diagnostic and therapeutic dilemmas during management.
A recent Indian expert group meeting strongly recommended the incorporation of screening for CAH in Indian newborn screening programs [6]. The advantages of screening include prevention of salt wasting crisis, prevention of hypoglycaemia, reduced mortality, reduced duration of hospitalizations, and reduced period of incorrect sex assignment [7]. There are data from Indian centres to support the utility of neonatal screening in our settings [6, 7]. The endocrine society guidelines [1, 5] have given an option to clinicians regarding growth-promoting therapy for children with CAH with predicted adult height below < –2.25 SD. It has been observed that children with CAH do not have advanced bone age in the first 2 years of life, despite elevated androgen levels [8]. The endocrine society recommends annual bone age assessment in children with CAH above 2 years. Bone age has significant utility in the diagnosis as well as monitoring the adequacy of therapy in CAH.
The guidelines also stressed the utility of regular periodic follow-up and the importance of stress dosing. The judicious and rationale use of dual-energy X-ray absorptiometry (DEXA) scan, adrenal imaging, and ACTH stimulation test has also been emphasized [1, 5]. Optimal steroid replacement with close, meticulous monitoring for features of glucocorticoid excess and androgenic excess states is recommended [5]. Faltering of growth is often the first sign of glucocorticoid excess state. The guidelines recommend judicious usage of steroid replacement in non-classical CAH [5].
What is new in the guidelines? The initial guidelines from the Endocrine Society recommended that children with severely virilized genitalia (Prader 3) should undergo neurovascular sparing clitoroplasty, vaginoplasty using urogenital mobilization, and perineal reconstruction [1]. Proponents of early surgery put forth better cosmetic results, patient preference, parent preference, and lowered complication rates as arguments in favour of early surgery [9, 10]. The more recent guidelines adopted a conservative approach [5]. They recommended that minimally virilized girls be observed and surgery delayed until the child is older. The new guidelines also recommend that caregivers provide the option of delaying surgery and/or observing in all girls with CAH. Early surgery to repair urogenital sinus could be considered in those with severely virilized genitalia.
In view of the modifications in the guidelines on the evaluation and management of children with CAH, we decided to perform this audit of children and adolescents with CAH as per the guidelines of the Endocrine Society.
Material and methods
A case record review of electronic maintenance records of 35 children aged 0-18 years with a diagnosis of CAH who have been under treatment and follow-up in the paediatric endocrinology clinic of a tertiary care referral hospital in South India from January 2014 to November 2021 was performed. Children with CAH were followed up periodically in a multidisciplinary clinic consisting of a paediatric endocrinologist, neonatologist, paediatric surgeon, and an ophthalmologist. Diagnosis of CAH was established based on elevated 17-hydroxyprogesterone (17 OHP) levels (either basal or ACTH stimulated) and features of androgen excess. Neonatal screening for CAH is done by heel prick technique at 72 hours of life on filter paper by a trained technician using competitive enzyme linked immunosorbent assay (ELISA) technique. Blood 17-OHP values are considered borderline between 12.3 and 30 ng/ml and positive beyond 30 ng/ml [7]. Children were classified on the basis of age of onset, and clinical and biochemical presentation into one of the three types of CAH – salt wasting, simple virilizing or non-classical CAH. Genital appearance was classified into one of the stages of Prader [11]. Children with glucocorticoid deficiency were administered oral hydrocortisone in a dose of 10–20 mg/m2, and those with mineralocorticoid deficiency were treated with oral fludrocortisone (0.05–0.3 mg). Infants with salt wasting were treated with oral salt (2–4 meq/kg/day of NaCl). The subjects with CAH were followed up in the endocrine clinic with growth monitoring, sexual maturity assessment as per Tanner’s staging, blood pressure, and biochemical measurement of 17 OHP. Bone age was assessed as clinically indicated using the Greulich Pyle atlas [12]. Final height prediction was ascertained based on Bayley Pinneau tables [13].
The management team focused on 10 major aspects of care of children with CAH and formulated 10 questions to audit pivotal aspects of care of children with CAH (screening, diagnosis, and medical and surgical management). The areas of focus include genital surgery, neonatal screening for CAH, stress dosing of CAH, need for ACTH stimulation test, growth-promoting therapy, bone-age assessment, adrenal imaging, bone mineral density assessment, adequacy of hormone replacement, and management of non-classical CAH. Questions to assess the management of CAH were formulated (Table I), and the diagnosis and management of children based on case records were scrutinized for data, and the results were entered in a Microsoft Excel spreadsheet.
Table I
Questions in audit sheet
Results
The baseline clinical, biochemical, and radiological parameters of the study population are presented in Table II. The results of the audit are presented below (summarized in Fig. 1).
Table II
Clinical, biochemical and radiological parameters of study sample (n = 35)
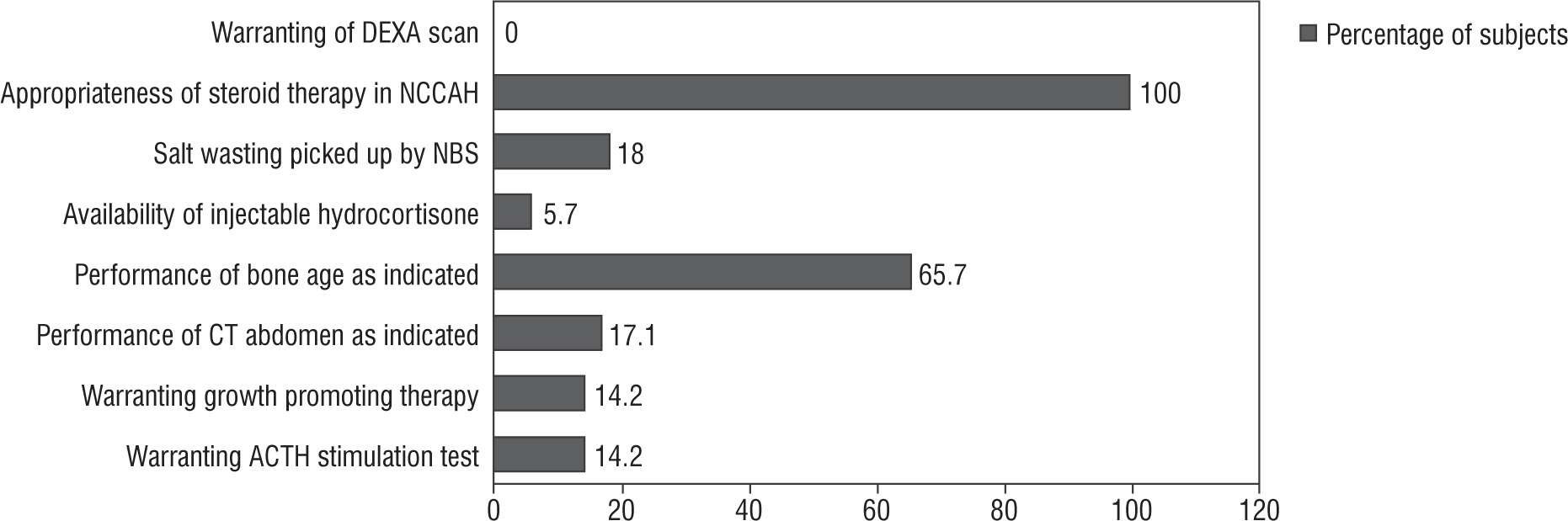
Screening: Four babies were identified on the basis of a neonatal screening program (2 females with virilization and 2 asymptomatic males). A possible salt wasting crisis was averted in all 4 positive-screened subjects owing to early hormone replacement.
Diagnosis: We observed that the height SD score of our study population was –0.12 ±1.6. The prevalence of short stature in our series (height SD score < –2) was 20%. We had one child with non-classical CAH, who presented with pubarche and was confirmed on ACTH stimulation test. She was initiated on oral hydrocortisone at a dose of 10 mg/m2 because she had advanced bone age. None of the children developed fractures; hence, none of them underwent a DEXA scan. Inconclusive basal 17 OH progesterone levels warranted ACTH stimulation tests in 5 children. One child was diagnosed as non-classical CAH, and fou4r children were diagnosed as simple virilizing CAH. All of them were initiated on oral hydrocortisone. Bone age was performed in 23 out of 35 (65.7%) subjects, as clinically indicated. Bone age was advanced in 40% of cases. CT abdomen was performed in 6 children. The indication for imaging was appropriate (unexplained hypertension [n = 2], borderline 17 OHP and disproportionate androgen elevation [n = 2], one had antenatal adrenal cyst and one had disproportionately high androgen levels). CT of the abdomen revealed no tumour in all 6 cases.
Medical therapy: All children were advised on stress dosing of steroids. Home injection of intramuscular hydrocortisone was provided to 2 children who presented with salt wasting crisis. Emergency usage of intramuscular hydroco rtisone was warranted in one child. Five (14%) children (3 males and 2 females) received growth-promoting therapy with gonadotropin-releasing hormone (GnRH) analogue therapy as indicated by low predicted final adult height (< 160 cm in males and < 150 cm in females). Three of them had precocious puberty. The median duration of therapy was 1.6 years. Chronological age and bone age at initiation of therapy were 7.6 ±2.5 years and 10.3 ±2.3 years, respectively. They were initiated on a 3-monthly pharmacological depot preparation of Leupride (Sunpharma) 11.25 mg at a frequency of 10–12 weeks [14]. The median baseline and endline predicted adult height were 145 cm and 152 cm, respectively. None received growth hormone therapy.
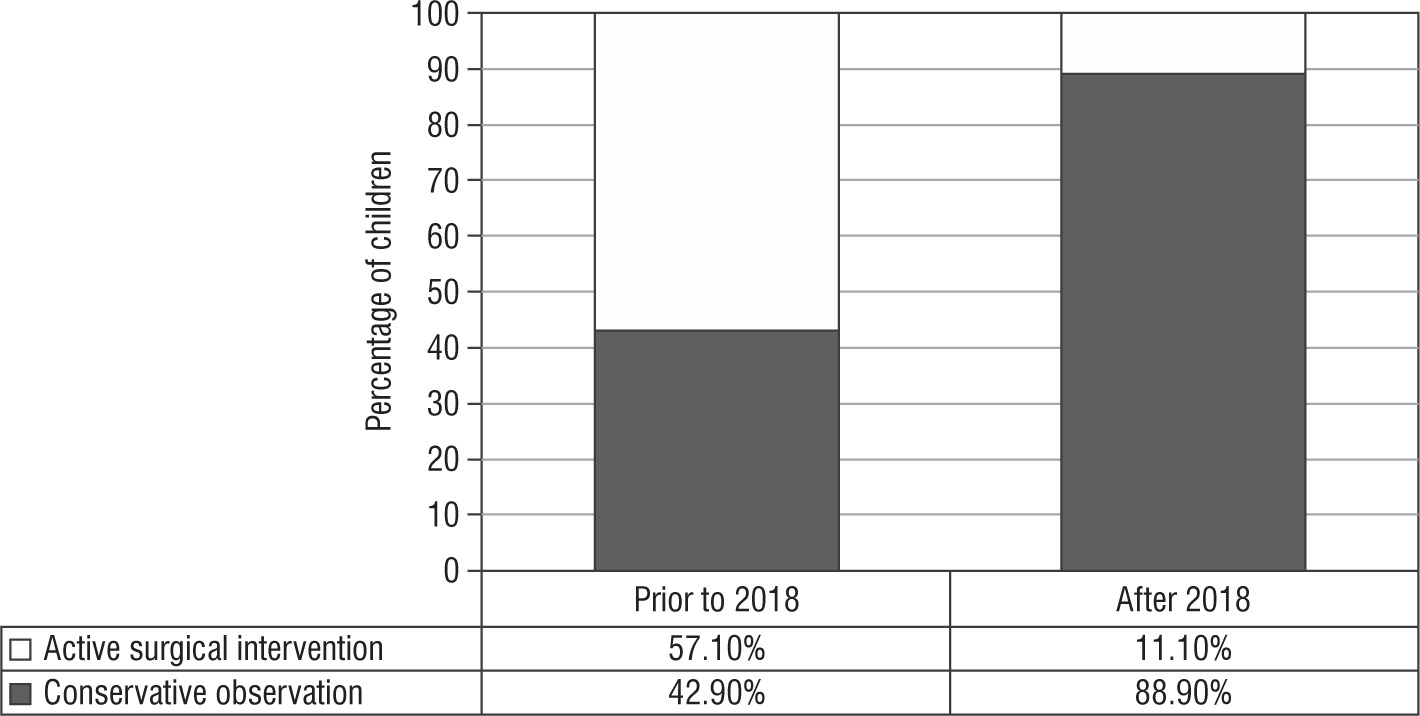
Surgical management: Prior to the 2018 guidelines, 4 out of 7 girls (of whom 5 [71.4%] had Prader stage 3 or more) underwent feminizing genitoplasty (57.1%) at a median age of surgery of 0.8 ±2.1 years. After the revision of the guideline, surgical management was performed in one out of 9 cases indicated (11.1%), of which 7 (77.7%) had Prader stage 3 or more of virilisation (Fig. 2). This child had severely virilized genitalia with a clitoral length of 3 cm and urogenital sinus resulting in recurrent urinary infection, and hence underwent genitoplasty at 5 years of age because the changes persisted despite good biochemical control of androgens.
Discussion
In our series, we observed that neonatal screening was able to identify 4 (11%) children with CAH, averting a possible salt wasting crisis in all 4 children including 2 asymptomatic males who were treated with oral hydrocortisone and fludrocortisone prior to the onset of symptoms, highlighting the utility of CAH screening as a part of the newborn screen. A study from Sweden has established that the incidence of CAH increases with the incorporation of screening (from 1 in 18,600 prior to screening to 1 in 12,800 after incorporation of screening) [15]. A study from Australia also demonstrated an earlier age at diagnosis in those screened (median day 13 in screened versus day 16 in unscreened) [16]. Data on screen-positive CAH is reported to be 1 in 2036, 1 in 9983, 1 in 2600, and 1 in 2800 newborns from various parts of our country [2–4]. Screening for CAH is currently not universal in our country. Many units are adopting the screening program. Thus, hitherto undiagnosed male CAH babies were identified prior to onset of salt wasting crisis, reinforcing the benefit of screening all babies for CAH.
We observed that the height SD score of our study population was –0.12 ±1.6 and short stature in 20%. A study from North India described the prevalence of short stature as 31% and low growth velocity in 55.6% [17]. The mean height SDS was described as –0.6. A study from western India described the faltering of height SDS from –0.1 to –0.3 and –0.4 to –0.5 in boys and girls with CAH, respectively [18]. They described the final height Z-score in a subset at –1.8 in salt wasting CAH and –1.6 in simple virilizing CAH. Thus, our study reinforces growth monitoring and optimal steroid doses in the treatment of CAH. Adrenal androgens vial local aromatization lead to advancement of skeletal maturity and compromised bone age. Annual bone age assessment is pivotal to ascertain control and optimize final height. Forty per cent of our subjects who had bone age assessed had advanced bone age.
In our study, 5 children received GnRH analogue therapy resulting in an improved predicted adult height of 7 cm. A study on 12 children from Turkey with central precocious puberty due to CAH initiated on GnRH analogue therapy at a chronological age of 6.8 years and bone age of 11 years resulted in an improvement in predicted adult height of 5 cm [19]. An Indian study from North India involved 5 subjects treated with GnRH analogue therapy, which resulted in improved predicted adult height by 8.2 cm [20]. It is pivotal that the initiation of GnRH analogue therapy is individualized for every case. Height prediction using Bayley Pinneau tables has recently been reported to be accurate for Indian children with CAH [18]. These children need to be followed up until they reach their adult height.
Treatment of non-classical CAH as per guidelines is indicated in children with significant advancement of bone age or overt virilization. Non-classical CAH is rare in children. A study from Eastern India observed that the median age of non-classical CAH was 20.4 years [21]. Oral hydrocortisone is indicated in children with: prepubertal growth acceleration, advancement of bone age, and stimulated cortisol < 18 µg/dl [22]. One child in our series had non-classical CAH and was treated with oral hydrocortisone in view of advanced bone age. To alleviate the risk of hypoglycaemia and dyselectrolytaemia, acute adrenal insufficiency should be prevented in every child with CAH on therapy. In our audit, one child warranted home emergency hydrocortisone injection, which was given at the crucial time, and an acute crisis was averted. Patients should have access to parenteral hydrocortisone and be trained to administer parenteral hydrocortisone in emergency situations [5]. Single, high, emergency doses of hydrocortisone do not worsen the growth prognosis.
In our series, after the 2018 guidelines, a conservative surgical management approach was adopted in 88.9% of cases versus 42.9% prior to the guidelines. Long-term follow-up studies have described reduced clitoral sensitivity, which is linearly related to difficulty in sexual function [23, 24]. Also, significant shortened vagina, higher sexual dysfunction, and sexual distress are described in these subjects on long-term follow-up [25]. Deferment of surgery until adolescence as the functional utility of vagina arises post childhood because the child is emotionally mature enough to cope, and endogenous oestrogen facilitates a favourable result [26]. A collective decision by a multidisciplinary team along with the caregivers is taken on the timing of surgery. A conscious decision to postpone surgery may be an active decision in the interest of patient autonomy. These children need to be followed up, and the change in their quality of life needs to be ascertained on follow-up.
Our study is not without limitations. The subjects picked up on neonatal screening include both inborn deliveries as well as babies screened at other centres and referred to our centre owing to a positive result. Thus, we are unable to present the pickup rate of neonatal screening. The small sample size is also a limitation of our study. These subjects need to be followed up to ascertain changes in quality of life and attainment of final height.
To conclude, we conducted an audit of 35 children with CAH. A shift to conservative surgical management of females, utility of neonatal screening for CAH, and judicious use of growth promoting therapy was observed, and their importance is highlighted. The need for bone age testing and emergency hydrocortisone provision is warranted.









