
28.06.2022
Redaktor:
Aleksandra Lang
Stwardnienie rozsiane pierwotnie postępujące – droga pacjenta od lekarza rodzinnego do neurologa
SM jest chorobą złożoną. Wiele genów zwiększa podatność na zachorowanie przy obecności określonych czynników środowiskowych, zwłaszcza: obniżonego poziomu witaminy D we krwi lub zmniejszonej ekspozycji na promieniowanie B światła ultrafioletowego (UVB), przebytego zakażenia wirusem Epsteina-Barr (EBV), otyłości czy palenia papierosów.
Autor: Anna Jamroz-Wiśniewska
Wstęp
Postać pierwotnie postępująca występuje w 10–15% przypadków stwardnienia rozsianego (sclerosis multiplex – SM), które jest autoimmunologiczną chorobą demielinizacyjną ośrodkowego układu nerwowego i prowadzi do zapalenia, a następnie zwyrodnienia w mózgu i rdzeniu kręgowym oraz nieodwracalnej utraty aksonów. Stwardnienie rozsiane jest najczęstszą przyczyną nieurazowej niepełnosprawności u młodych dorosłych. Częstość występowania SM na świecie wzrasta – w 2020 r. 2,8 mln ludzi na świecie było dotkniętych chorobą, a w 2013 r. ok. 30% mniej. W Polsce, która jest krajem o wysokim ryzyku zachorowania na SM, jest ok. 45 000–50 000 chorych.
Stwardnienie rozsiane jest chorobą złożoną. Wiele genów zwiększa podatność na zachorowanie przy obecności określonych czynników środowiskowych, zwłaszcza: obniżonego poziomu witaminy D we krwi lub zmniejszonej ekspozycji na promieniowanie B światła ultrafioletowego (UVB), przebytego zakażenia wirusem Epsteina-Barr (EBV), otyłości czy palenia papierosów [1, 2].
Choroba przebiega w podstawowych postaciach klinicznych – rzutowo-remisyjnej (relapsing-remitting SM – RR-SM) i postępującej. Postać postępująca może następować po początkowej fazie RR-SM, wówczas nazywa się ją postacią wtórnie postępującą, albo od początku mieć taki charakter, wtedy mówimy o postaci pierwotnie postępującej (primary progressive SM – PP-SM) [1, 3, 4].
Rozpoznanie postaci PP-SM
Postać PP-SM różni się od częściej występującej i lepiej rozpoznawalnej postaci RR-SM przebiegiem i zmianami neuropatologicznymi, inne są też kryteria diagnostyczne. Rokowanie jest gorsze, a progresja choroby szybsza. Postać PP-SM często przez długi czas pozostaje nierozpoznana. Charakteryzuje się stopniowym narastaniem niepełnosprawności od początku rozwoju objawów, nie występują rzuty choroby ani okresy remisji typowe dla postaci RR-SM. Najczęstszą prezentacją kliniczną jest postępujący spastyczny niedowład kończyn dolnych z towarzyszącymi zaburzeniami zwieraczy, ale mogą wystąpić również niezborność lub ataksja (czuciowa, móżdżkowa), zaburzenia poznawcze, postępujące upośledzenie widzenia. Zwykle w ciągu kilku lat pacjent potrzebuje przy chodzeniu pomocy drugiej osoby, kul łokciowych czy podpórki, a niedługo potem jest zmuszony do korzystania z wózka inwalidzkiego [1, 3, 4].
Pacjent, diagnozowany później jako chory na PP-SM, najczęściej ponad 40-letni, zwykle zgłasza narastające od wielu miesięcy osłabienie kończyn dolnych. Kiedyś biegał amatorsko, a ostatnio już nie biega, nie może nawet podbiec do autobusu. Jest ogólnie osłabiony i nadmiernie zmęczony. Zaczął też mieć problemy z nietrzymaniem moczu i impotencją. W takim przypadku w diagnostyce różnicowej należy z pewnością wziąć pod uwagę PP-SM.
Na podstawie kryteriów McDonalda z 2017 r. diagnozę PP-SM stawia się u pacjentów z:
• roczną progresją niepełnosprawności (określaną retrospektywnie lub prospektywnie)
• plus 2 z następujących:
• obecność 1 lub więcej zmian hiperintensywnych w obrazach T2-zależnych w badaniu rezonansu magnetycznego (magnetic resonance – MR) charakterystycznych dla SM w 1 lub więcej obszarach mózgu: okołokomorowo, korowo/podkorowo lub podnamiotowo,
• obecność 2 lub więcej zmian hiperintensywnych w obrazach T2-zależnych w badaniu MR w rdzeniu kręgowym,
• obecność prążków oligoklonalnych (białka oligoklonalnego) w płynie mózgowo-rdzeniowym.
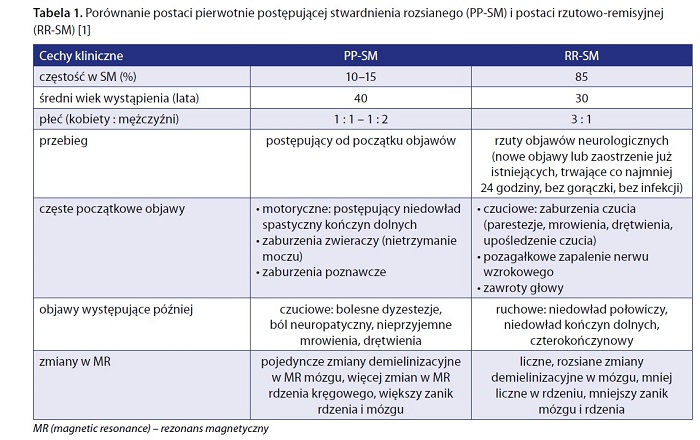
Pacjenci z PP-SM są nieco starsi w porównaniu z pacjentami z RR-SM (średni wiek to 40 lat). Postać PP-SM występuje z porównywalną częstością u obu płci (z niewielką przewagą mężczyzn). U pacjentów z PP-SM dominują początkowo objawy ruchowe, o charakterze rdzeniowym, zaburzenia zwieraczy, a dopiero potem dołączają się zaburzenia czucia. W postaci RR-SM jest odwrotnie – początkowo pacjenci skarżą się na parestezje, zaburzenia czucia, mogą przebyć pozagałkowe zapalenie nerwu wzrokowego, a zwykle dopiero po co najmniej kilku latach trwania choroby rozwijają się objawy motoryczne (niedowłady). W badaniach MR w przypadku PP-SM stwierdza się pojedyncze zmiany w mózgu, więcej zmian w rdzeniu kręgowym i większą atrofię rdzenia i mózgu w porównaniu z RR-SM [1, 3] (tab. 1).
Pacjent z podejrzeniem postaci PP-SM powinien przejść gruntowną diagnostykę różnicową. Należy wykluczyć ucisk rdzenia kręgowego (przepuklina jądra miażdżystego, guz, jamistość rdzenia), postępującą mielopatię metaboliczną (m.in. niedobór witaminy B12), przyczyny genetyczne (dziedziczna parapareza spastyczna, ataksja rdzeniowo-móżdżkowa, leukodystrofie), choroby infekcyjne (m.in. HIV, kiła, neuroborelioza) [1].
Stwierdzenie postępujących objawów neurologicznych, najczęściej – jak już wspomniano – niedowładu kończyn dolnych, jest wskazaniem do skierowania pacjenta do poradni neurologicznej, najlepiej w ośrodku zajmującym się leczeniem SM. W praktyce pacjent często pozostaje w diagnostyce POZ, jest kierowany do ortopedy, chirurga czy do neurologa poza ośrodkiem leczącym SM, co wydłuża ustalenie rozpoznania. Stwierdzono, że od wystąpienia pierwszych objawów choroby do właściwego rozpoznania u pacjentów z PP-SM upływa w Polsce ok. 6 lat, dwukrotnie więcej niż w przypadku postaci RR-SM [4].
Ostatnio w Klinice Neurologii w Lublinie hospitalizowano 62-letniego mężczyznę z wywiadem postępujących objawów neurologicznych od ok. 10 lat. W tym czasie, zanim rozpoznano PP-SM, z powodu podejrzenia mielopatii szyjnej i zespołu ogona końskiego przebył dwie operacje dyskopatii – kręgosłupa lędźwiowo-krzyżowego i szyjnego, bez poprawy. Obecnie miał głęboki niedowład spastyczny kończyn dolnych i objawy pęcherza neurogennego (nietrzymanie moczu), poruszał się na dłuższe dystanse z pomocą wózka inwalidzkiego, a w przeszłości trenował judo… Spełniał wymogi programu lekowego NFZ i został włączony do leczenia okrelizumabem.
Leczenie PP-SM – okrelizumab
Pierwszym i jedynym lekiem, który wykazał skuteczność w leczeniu PP-SM jest właśnie okrelizumab (preparat Ocrevus). Od listopada 2019 r. jest refundowany w ramach programu NFZ. To humanizowane przeciwciało monoklonalne skierowane przeciwko antygenom CD20 na komórkach B w postaci PP-SM opóźnia narastanie niepełnosprawności o 24% vs placebo (badanie ORATORIO). W obserwacji 6,5-letniej okrelizumab zmniejszał o 42% ryzyko progresji niepełnosprawności do potrzeby stosowania wózka inwalidzkiego u pacjentów od początku stosujących ten lek. Stwierdzono, że opóźnia o 7,1 roku czas do potrzeby stosowania wózka inwalidzkiego. Zapobiega powstawaniu nowych zmian demielinizacyjnych widocznych w badaniu MR oraz progresji zaniku mózgu. Okrelizumab przeciwdziała rozwojowi niepełnosprawności kończyn górnych i dolnych, powoduje utrzymanie niezależności i lepszej jakości życia. Wykazano też skuteczność tego leku w postaci RR-SM (jest refundowany również w tej postaci). Lek jest podawany dożylnie raz na pół roku, co jest wygodne dla pacjentów i przyczynia się do dobrej adherencji. Tolerancja leczenia jest dobra, mogą wystąpić objawy związane z infuzją i infekcje (najczęściej łagodne do umiarkowanych) [6].
Zgodnie z treścią programu NFZ do leczenia okrelizumabem kwalifikowani są pacjenci z PP-SM, u których stwierdza się aktywność w badaniu MR z ostatnich 12 miesięcy (co najmniej jedna nowa lub powiększająca się zmiana demielinizacyjna w obrazach T2-zależnych lub co najmniej jedna zmiana wzmacniająca się po podaniu kontrastu gadolinowego w obrazach T1-zależnych w porównaniu z badaniem poprzednim). Przed włączeniem pacjenta wymagane są: badanie RTG klatki piersiowej, badania przesiewowe w kierunku raka piersi u kobiet, badania przesiewowe w kierunku WZW typu B (może ulec reaktywacji) i podstawowe badania laboratoryjne [7].
Podsumowanie
Pacjenta z podejrzeniem postaci PP-SM należy jak najszybciej skierować do neurologa, optymalnie w ośrodku zajmującym się leczeniem SM, gdzie można ustalić ostateczne rozpoznanie i podjąć właściwe leczenie, a tym samym zapobiegać rozwojowi niepełnosprawności, utrzymać niezależność i lepszą jakość życia. Wcześniejsze rozpoczęcie leczenia powoduje dłuższe utrzymanie sprawności kończyn górnych i dolnych w porównaniu z pacjentami, u których terapia została włączona z opóźnieniem, dlatego tak ważna jest szybka i prawidłowa diagnoza [6].
Piśmiennictwo
1. Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol 2019; 26: 27-40.
2. Walton C, King R, Rechtman L i wsp. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler 2020; 26: 1816-1821.
3. Rice CM, Cottrell D, Wilkins A i wsp. Primary progressive multiple sclerosis: progress and challenges. J Neurol Neurosurg Psychiatry 2013; 84: 1100-1106.
4. Brola W, Sobolewski P, Żak M i wsp. Profile of Polish patients with primary progressive multiple sclerosis. Mult Scler Relat Disord 2019; 33: 33-38.
5. Thompson AJ, Banwell BL, Barkhof F i wsp. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17: 162-173.
6. Lamb YN. Ocrelizumab: a review in multiple sclerosis. Drugs 2022; 82: 323-334.
7. Obwieszczenie Ministra Zdrowia z dnia 24 sierpnia 2020 r. w sprawie wykazu refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych na dzień 1 września 2020 r. (DZ. URZ. Min. Zdr. 2020.60).
Artykuł opublikowany dzięki współpracy z firmą Roche Polska Sp. z o.o.
Wstęp
Postać pierwotnie postępująca występuje w 10–15% przypadków stwardnienia rozsianego (sclerosis multiplex – SM), które jest autoimmunologiczną chorobą demielinizacyjną ośrodkowego układu nerwowego i prowadzi do zapalenia, a następnie zwyrodnienia w mózgu i rdzeniu kręgowym oraz nieodwracalnej utraty aksonów. Stwardnienie rozsiane jest najczęstszą przyczyną nieurazowej niepełnosprawności u młodych dorosłych. Częstość występowania SM na świecie wzrasta – w 2020 r. 2,8 mln ludzi na świecie było dotkniętych chorobą, a w 2013 r. ok. 30% mniej. W Polsce, która jest krajem o wysokim ryzyku zachorowania na SM, jest ok. 45 000–50 000 chorych.
Stwardnienie rozsiane jest chorobą złożoną. Wiele genów zwiększa podatność na zachorowanie przy obecności określonych czynników środowiskowych, zwłaszcza: obniżonego poziomu witaminy D we krwi lub zmniejszonej ekspozycji na promieniowanie B światła ultrafioletowego (UVB), przebytego zakażenia wirusem Epsteina-Barr (EBV), otyłości czy palenia papierosów [1, 2].
Choroba przebiega w podstawowych postaciach klinicznych – rzutowo-remisyjnej (relapsing-remitting SM – RR-SM) i postępującej. Postać postępująca może następować po początkowej fazie RR-SM, wówczas nazywa się ją postacią wtórnie postępującą, albo od początku mieć taki charakter, wtedy mówimy o postaci pierwotnie postępującej (primary progressive SM – PP-SM) [1, 3, 4].
Rozpoznanie postaci PP-SM
Postać PP-SM różni się od częściej występującej i lepiej rozpoznawalnej postaci RR-SM przebiegiem i zmianami neuropatologicznymi, inne są też kryteria diagnostyczne. Rokowanie jest gorsze, a progresja choroby szybsza. Postać PP-SM często przez długi czas pozostaje nierozpoznana. Charakteryzuje się stopniowym narastaniem niepełnosprawności od początku rozwoju objawów, nie występują rzuty choroby ani okresy remisji typowe dla postaci RR-SM. Najczęstszą prezentacją kliniczną jest postępujący spastyczny niedowład kończyn dolnych z towarzyszącymi zaburzeniami zwieraczy, ale mogą wystąpić również niezborność lub ataksja (czuciowa, móżdżkowa), zaburzenia poznawcze, postępujące upośledzenie widzenia. Zwykle w ciągu kilku lat pacjent potrzebuje przy chodzeniu pomocy drugiej osoby, kul łokciowych czy podpórki, a niedługo potem jest zmuszony do korzystania z wózka inwalidzkiego [1, 3, 4].
Pacjent, diagnozowany później jako chory na PP-SM, najczęściej ponad 40-letni, zwykle zgłasza narastające od wielu miesięcy osłabienie kończyn dolnych. Kiedyś biegał amatorsko, a ostatnio już nie biega, nie może nawet podbiec do autobusu. Jest ogólnie osłabiony i nadmiernie zmęczony. Zaczął też mieć problemy z nietrzymaniem moczu i impotencją. W takim przypadku w diagnostyce różnicowej należy z pewnością wziąć pod uwagę PP-SM.
Na podstawie kryteriów McDonalda z 2017 r. diagnozę PP-SM stawia się u pacjentów z:
• roczną progresją niepełnosprawności (określaną retrospektywnie lub prospektywnie)
• plus 2 z następujących:
• obecność 1 lub więcej zmian hiperintensywnych w obrazach T2-zależnych w badaniu rezonansu magnetycznego (magnetic resonance – MR) charakterystycznych dla SM w 1 lub więcej obszarach mózgu: okołokomorowo, korowo/podkorowo lub podnamiotowo,
• obecność 2 lub więcej zmian hiperintensywnych w obrazach T2-zależnych w badaniu MR w rdzeniu kręgowym,
• obecność prążków oligoklonalnych (białka oligoklonalnego) w płynie mózgowo-rdzeniowym.
Pacjenci z PP-SM są nieco starsi w porównaniu z pacjentami z RR-SM (średni wiek to 40 lat). Postać PP-SM występuje z porównywalną częstością u obu płci (z niewielką przewagą mężczyzn). U pacjentów z PP-SM dominują początkowo objawy ruchowe, o charakterze rdzeniowym, zaburzenia zwieraczy, a dopiero potem dołączają się zaburzenia czucia. W postaci RR-SM jest odwrotnie – początkowo pacjenci skarżą się na parestezje, zaburzenia czucia, mogą przebyć pozagałkowe zapalenie nerwu wzrokowego, a zwykle dopiero po co najmniej kilku latach trwania choroby rozwijają się objawy motoryczne (niedowłady). W badaniach MR w przypadku PP-SM stwierdza się pojedyncze zmiany w mózgu, więcej zmian w rdzeniu kręgowym i większą atrofię rdzenia i mózgu w porównaniu z RR-SM [1, 3] (tab. 1).
Pacjent z podejrzeniem postaci PP-SM powinien przejść gruntowną diagnostykę różnicową. Należy wykluczyć ucisk rdzenia kręgowego (przepuklina jądra miażdżystego, guz, jamistość rdzenia), postępującą mielopatię metaboliczną (m.in. niedobór witaminy B12), przyczyny genetyczne (dziedziczna parapareza spastyczna, ataksja rdzeniowo-móżdżkowa, leukodystrofie), choroby infekcyjne (m.in. HIV, kiła, neuroborelioza) [1].
Stwierdzenie postępujących objawów neurologicznych, najczęściej – jak już wspomniano – niedowładu kończyn dolnych, jest wskazaniem do skierowania pacjenta do poradni neurologicznej, najlepiej w ośrodku zajmującym się leczeniem SM. W praktyce pacjent często pozostaje w diagnostyce POZ, jest kierowany do ortopedy, chirurga czy do neurologa poza ośrodkiem leczącym SM, co wydłuża ustalenie rozpoznania. Stwierdzono, że od wystąpienia pierwszych objawów choroby do właściwego rozpoznania u pacjentów z PP-SM upływa w Polsce ok. 6 lat, dwukrotnie więcej niż w przypadku postaci RR-SM [4].
Ostatnio w Klinice Neurologii w Lublinie hospitalizowano 62-letniego mężczyznę z wywiadem postępujących objawów neurologicznych od ok. 10 lat. W tym czasie, zanim rozpoznano PP-SM, z powodu podejrzenia mielopatii szyjnej i zespołu ogona końskiego przebył dwie operacje dyskopatii – kręgosłupa lędźwiowo-krzyżowego i szyjnego, bez poprawy. Obecnie miał głęboki niedowład spastyczny kończyn dolnych i objawy pęcherza neurogennego (nietrzymanie moczu), poruszał się na dłuższe dystanse z pomocą wózka inwalidzkiego, a w przeszłości trenował judo… Spełniał wymogi programu lekowego NFZ i został włączony do leczenia okrelizumabem.
Leczenie PP-SM – okrelizumab
Pierwszym i jedynym lekiem, który wykazał skuteczność w leczeniu PP-SM jest właśnie okrelizumab (preparat Ocrevus). Od listopada 2019 r. jest refundowany w ramach programu NFZ. To humanizowane przeciwciało monoklonalne skierowane przeciwko antygenom CD20 na komórkach B w postaci PP-SM opóźnia narastanie niepełnosprawności o 24% vs placebo (badanie ORATORIO). W obserwacji 6,5-letniej okrelizumab zmniejszał o 42% ryzyko progresji niepełnosprawności do potrzeby stosowania wózka inwalidzkiego u pacjentów od początku stosujących ten lek. Stwierdzono, że opóźnia o 7,1 roku czas do potrzeby stosowania wózka inwalidzkiego. Zapobiega powstawaniu nowych zmian demielinizacyjnych widocznych w badaniu MR oraz progresji zaniku mózgu. Okrelizumab przeciwdziała rozwojowi niepełnosprawności kończyn górnych i dolnych, powoduje utrzymanie niezależności i lepszej jakości życia. Wykazano też skuteczność tego leku w postaci RR-SM (jest refundowany również w tej postaci). Lek jest podawany dożylnie raz na pół roku, co jest wygodne dla pacjentów i przyczynia się do dobrej adherencji. Tolerancja leczenia jest dobra, mogą wystąpić objawy związane z infuzją i infekcje (najczęściej łagodne do umiarkowanych) [6].
Zgodnie z treścią programu NFZ do leczenia okrelizumabem kwalifikowani są pacjenci z PP-SM, u których stwierdza się aktywność w badaniu MR z ostatnich 12 miesięcy (co najmniej jedna nowa lub powiększająca się zmiana demielinizacyjna w obrazach T2-zależnych lub co najmniej jedna zmiana wzmacniająca się po podaniu kontrastu gadolinowego w obrazach T1-zależnych w porównaniu z badaniem poprzednim). Przed włączeniem pacjenta wymagane są: badanie RTG klatki piersiowej, badania przesiewowe w kierunku raka piersi u kobiet, badania przesiewowe w kierunku WZW typu B (może ulec reaktywacji) i podstawowe badania laboratoryjne [7].
Podsumowanie
Pacjenta z podejrzeniem postaci PP-SM należy jak najszybciej skierować do neurologa, optymalnie w ośrodku zajmującym się leczeniem SM, gdzie można ustalić ostateczne rozpoznanie i podjąć właściwe leczenie, a tym samym zapobiegać rozwojowi niepełnosprawności, utrzymać niezależność i lepszą jakość życia. Wcześniejsze rozpoczęcie leczenia powoduje dłuższe utrzymanie sprawności kończyn górnych i dolnych w porównaniu z pacjentami, u których terapia została włączona z opóźnieniem, dlatego tak ważna jest szybka i prawidłowa diagnoza [6].
Piśmiennictwo
1. Dobson R, Giovannoni G. Multiple sclerosis – a review. Eur J Neurol 2019; 26: 27-40.
2. Walton C, King R, Rechtman L i wsp. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler 2020; 26: 1816-1821.
3. Rice CM, Cottrell D, Wilkins A i wsp. Primary progressive multiple sclerosis: progress and challenges. J Neurol Neurosurg Psychiatry 2013; 84: 1100-1106.
4. Brola W, Sobolewski P, Żak M i wsp. Profile of Polish patients with primary progressive multiple sclerosis. Mult Scler Relat Disord 2019; 33: 33-38.
5. Thompson AJ, Banwell BL, Barkhof F i wsp. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17: 162-173.
6. Lamb YN. Ocrelizumab: a review in multiple sclerosis. Drugs 2022; 82: 323-334.
7. Obwieszczenie Ministra Zdrowia z dnia 24 sierpnia 2020 r. w sprawie wykazu refundowanych leków, środków spożywczych specjalnego przeznaczenia żywieniowego oraz wyrobów medycznych na dzień 1 września 2020 r. (DZ. URZ. Min. Zdr. 2020.60).
Artykuł opublikowany dzięki współpracy z firmą Roche Polska Sp. z o.o.





