
Introduction
Familial hypercholesterolaemia (FH) is an inherited disorder of plasma lipoprotein metabolism characterised by markedly increased and sustained plasma low-density lipoprotein cholesterol (LDL-C) concentrations, which increase the risk of premature atherosclerotic cardiovascular disease (CVD) in the affected individuals by 3- to 10-fold versus the general population [1]. Of the two FH types, heterozygous FH (HeFH) is milder but more frequent, with a prevalence of 1/500 to 1/200, whereas the homozygous form (HoFH) is severe but less prevalent (about 1/1,000,000) [1]. The HoFH is caused by mutations in the genes for either LDL Receptor (LDLR) protein, apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), or LDL adaptor protein (LDLRAP) [2]. About 85% of patients with HoFH have mutations in the LDLR gene, located on chromosome 19. The LDLR is a transmembrane protein on the surface of hepatocytes, which mediates the uptake of plasma LDL into hepatocytes for further metabolism. Mutations in the LDLR gene either reduce the number or function of LDLR molecules, which results in decreased clearance of LDL. As a consequence, the plasma concentrations LDL as well as total cholesterol (TC) increase. The plasma concentrations of lipoprotein particles, predominantly LDL, are critical determinants for initiating changes in the vascular endothelium, which result in atherosclerosis and premature CAD as well as valvular lesions and peripheral arterial disease [1, 2].
Despite a grave prognosis, FH remains underdiagnosed and undertreated; only about 20% of patients are diagnosed during childhood. The clinical diagnosis is usually based on one of the several proposed criteria, such as the Simon Broome Register Group criteria, Dutch Lipid Clinic Network criteria, Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria, the Japanese criteria, and the Simplified Chinese Criteria for Familial Hypercholesterolaemia (SCCFH) [2, 3]. However, the clinical diagnostic criteria for FH can be inaccurate and often need to be supported by genetic diagnosis for several reasons. The characterisation of genetic defect allows cascade testing, has prognostic utility and allows determination of response to lipid lowering drugs [2]. For example, the homozygous patients show higher basal LDL-C concentrations and a poorer response to therapy compared with compound heterozygotes [2]. The response to statins is limited in patients with homozygous or compound heterozygous for damaging or null LDLR variants because these drugs remove circulating LDL and remnant lipoproteins by increasing the LDLR expression on the cell surface [4]. The statins may exacerbate the disease in cases of FH caused by lysosomal acid lipase deficiency by increasing the endocytosis of cholesteryl esters. Drugs that do not require a competent LDLR for their action such as bile acid ion-exchange resins, niacin, ezetimibe, and lomitapide are effective in FH due to mutations in ATP binding cassette subfamily G [4]. Lastly, molecular diagnosis of HoFH may help to avoid situations where xanthoma is misdiagnosed as other skin lesions and therefore mistreated in clinical practice [5, 6].
In many developed countries, genetic screening is routinely performed in families with FH, especially HoFH, which has helped in the characterisation of prevalent mutations in their populations and allowed the development of cost-effective diagnostic evaluation [7, 8]. In India, however, children with FH are usually diagnosed and treated based on clinical criteria, and there are only limited data on the genetic diagnosis of FH [9]. One of the three previous genetic studies in children with HoFH was conducted on a patient population from Southwest India [10]. The other two studies from Northwest India observed that the spectrum of mutations in the Asian Indian population was quite heterogeneous [11, 12]. With India being a multi-ethnic country, wide variations are expected in the genetic mutations affecting patient populations in different regions of the country [13]. We therefore planned to determine the spectrum of mutations in the LDLR gene in our cohort of North Indian children with a clinical diagnosis of HoFH, and their parents.
Material and Methods
Study population
The probands (n = 8) were children of either sex, aged less than 16 years, and diagnosed clinically with HoFH using the Simon Broome criteria – briefly, presence of cutaneous or tendinous xanthomas, and/or plasma TC and LDL-C concentrations > 260 mg/dl (6.7 mmol/l) and > 155 mg/dl (4.0 mmol/l), respectively. Two family members of the probands, mainly parents also participated in the study. The study participants gave written informed consent prior to their enrolment, which adhered to the World Medical Association Declaration of Helsinki. Children were included with parental consent and their own assent wherever applicable. The study was approved by the Institute’s Ethics Committee (INT/IEC/2020/000064).
Biochemical analyses, and extraction, amplification, and sequencing of genomic DNA
After overnight fasting, blood from the study participants was obtained in tubes containing 0.1% ethylenediaminetetraacetic acid, and plasma was separated. Measurement of TC, high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) was done on an automated chemical analyser (Advia 1800 clinical chemistry analyser, Siemens Healthcare Diagnostics, USA). The LDL-C fraction was calculated by Friedewald equation as: LDL-C (mg/dl) = TC – HDL-C – (TG/5). Genomic DNA was extracted from blood using a QIAmp DNA blood kit (Qiagen). Specific primers for the 18 exons of the LDLR gene (RefSeq Accession: NG_00060) were designed using the Primer3www.bioinformatics.nl server. The primers designed in the adjoining intron regions flanked the whole exon. The extracted DNA samples were quantified using spectrophotometer (NanoDrop). The quality of DNA was checked based on the 260/280 absorption ratio, and the samples with ratios around 1.8 were selected for further amplification using PCR. The PCR amplicons were then purified using ExoSAP-IT (ThermoFisher Scientific) in a thermal cycler according to the manufacturer’s protocol. The amplicons and the primers were then submitted for Sanger Sequencing (Abi 3500 Genetic Analyzer) and the results were analysed using Automated Mutation Analysis Pipeline, and the reported variations were checked in the LDLR gene database (http://www.lovd.nl/LDLR). The pathogenicity of variants was analysed using bioinformatics software tools such as PolyPhen-2, SIFT, and Mutation Taster and Variant Effect Predictor (VEP) using plug-ins dbscSNV and MaxEntScan for splice site variants. Mean allele frequency was checked on the gnomAD database version 2.1.1. The variants were classified according to the American College of Medical Genetics guidelines based on a 5-tier system [14].
Statistical analysis
Data were analysed using Statistical Package for the Social Sciences (SPSS, version 23.0, IBM Corp., Armonk, NY, USA). Descriptive statistics were used for baseline comparison. The genotype phenotype relationship was determined by using univariate factors such as age, presence of xanthomas, LDL levels, and the gene variants. Spearman’s rank correlation coefficient was determined with each factor. The existence of Hardy-Weinberg equilibrium was analysed using gene frequencies obtained by simple gene counting, and the chi-square test with Yates’ correction was applied for comparing observed and expected values. A p-value of < 0.05 was considered statistically significant.
Results
Clinical characteristics of the study cohort
Eight families belonging to three major states of Northwest India, i.e. Punjab (5 families), Haryana (2 families), and Himachal Pradesh (1 family), were included in this study. Six probands were boys and 2 were girls; their mean age at diagnosis was 6.2 ±2.6 years (range 3–9 years). All patients except 2 presented with xanthomas. Family history of early CVD was not present in any patient; one parent was on treatment for elevated TC. The mean concentrations of TC, TG, and LDL-C were 548.7 ±180.7, 318.2 ±471.2, and 417.0 ±162.6 mg/dl, respectively. The mean carotid intima media thickness was 0.37±0.06 mm (normal reference values 0.34 ±0.05 mm, cut-off > 0.45 mm) [15].
Genetic analysis
A total of 50 variations were found in the 8 patients; 23 (46%) were non-coding, 19 (38%) synonymous, and 4 (8%) frameshift and missense variants. A majority of the variants (39, 78%) were single nucleotide variations (SNV), while 8 (16%) and 3 (6%) were deletions and insertions, respectively.
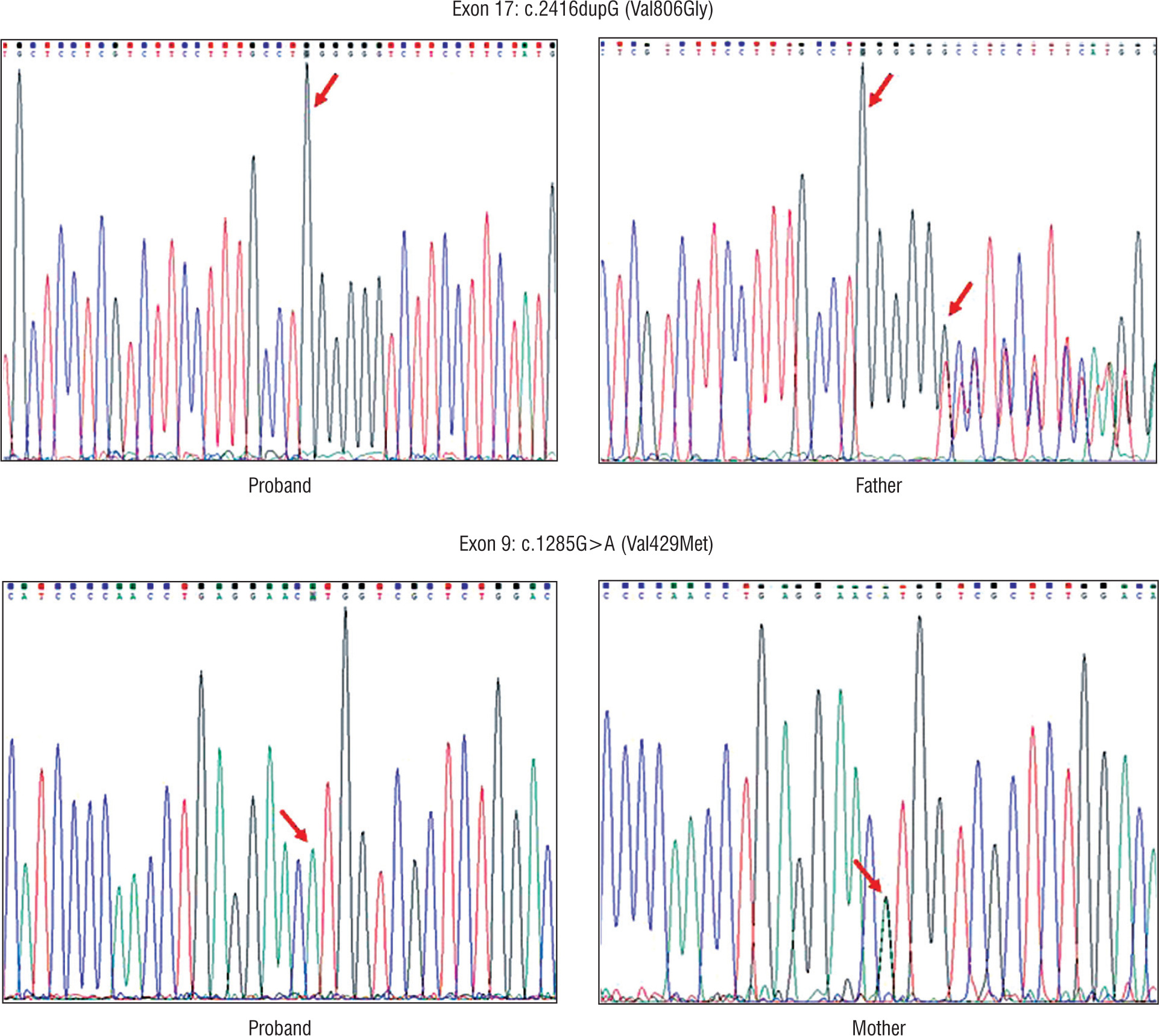
Four patients (50%) showed HoFH-causing variants in the LDLR gene (Table I). The other four did not show any clinically relevant mutation. Three out of four patients contained pathogenic variant (duplication) in exon 17 (c.2416dupG) creating an amino acid change at position 806 (p.Val806GlyfsTer11), leading to protein chain termination following 11 amino acids. The other variant (missense) was noted in exon 9 of the LDLR gene at position c.1285G>A. This mutation leads to the change in amino acid at position 429 (p.Val429Met) (Fig. 1). All the above-noted variants were tested in the parent’s or sibling’s DNA sample and were detected in heterozygous state in each case.
Table I
Pathogenic mutations in the LDLR gene detected in 4 patients and their corresponding lipid levels
Genotype-phenotype relationship
We did not find any relationship between the genotype and phenotype in our patients. The presence of pathogenic variants did not show any association to the age (χ2 = 0.0, p-value 1.0), presence of xanthoma (χ2 = 0.533, p-value 0.465), and concentrations of LDL-C (χ2 = 4.8, p-value 0.91) and TC (0.533, p-value 0.465).
Discussion
In our observational study, we characterised disease-causing mutations in the LDLR gene in 8 children with a definite or possible clinical diagnosis of HoFH. The pathogenic variants were found in 4 (50%) patients, 3 of whom had the same variant. In other cohorts of HoFH, LDLR gene mutations have been reported with a frequency ranging between 80 and almost 100% [1, 2, 11]. It is known that the range of LDLR variants changes between populations because approximately 1700 different mutations have been identified around the world (www.ucl.ac.uk/ldlr). Similarly, the proportion of true versus compound heterozygotes was 50% as compared to other cohorts that have very few true homozygotes [2].
The insertion observed in 3 patients at position 211 in exon 17 that resulted in duplication of G (c.2416dupG (p.Val806GlyfsTer11) causes the reading frame to shift at codon 806, changing valine to glycine and creating a premature stop codon [16]. This results in a truncated, abnormal protein product or loss of protein through nonsense-mediated mRNA decay. In one family, this variant was also found in a heterozygous state in the father, who had elevated LDL levels; the data for the mother could not be interpreted due to poor readings. In the second patient, the mother and a sibling (brother) were found to be heterozygous for the same mutation; father’s sample was not available for analysis. The sequencing data for the parents of the third child could be confirmed only in the mother, who had the same heterozygous variant. This mutation has been identified in patients from different ethnic backgrounds such as Japanese, Pakistani, and Indian [11, 17, 18].
Only one of our patients showed a mutation in exon 9, which is considered as one of the hot spots in studies performed on Asian Indians in South Africa [11]. This missense variant at position 11224052 results in replacement of valine with methionine at codon 429 of the LDLR protein (p.Val429Met) and causes a slower processing of the precursor LDLR protein to its mature form and hence its faster degradation [19]. The mother of the proband had the same heterozygous variant; however, it could not be identified in the father’s DNA. This variant has been reported in patients belonging to different ethnicities. It was found in 4 of 28 Greek patients, 2 of 1070 Italian individuals, and 161 members of a large Dutch family with FH [20–22].
We did not find any new variants in the LDLR gene in our cohort of HoFH children. However, the detected variants in our patients are different from previous studies from Northwest India, suggesting a different lineage or ancestry of our cohort despite belonging to a similar geographic location. This is explained by the genetic heterogeneity due to the multi-ethnic composition of the Indian population [13]. The limitations of our study were the small sample size, which was primarily due to financial constraints, and the inability to perform a robust mutation segregation analysis for all patients due to missing data of either parents or siblings.
In conclusion, the disease-causing variants in the LDLR gene were detected in only 50% of our patients with HoFH. Larger studies are recommended to characterise the variants in the genes other than the LDLR gene in patient populations with HoFH.









