
Introduction
Monogenic obesity is caused by a mutation in one of the single genes involved in the metabolic pathways controlling hunger and satiety [1]. The leptin-melanocortin pathway signalling to and from the hypothalamus is primarily involved in the regulation of hunger, satiety, and energy expenditure [2]. Leptin is a hormone secreted from the white adipose tissue and is a marker of energy reserves of the body. Leptin acts on its receptor (leptin receptor [LepR]), which is mainly expressed in the neurons in the hypothalamus. The binding of leptin to its receptor activates various intracellular pathways including Janus kinase 2 (JAK2) and signal transducer and activator of transcription (STAT) 3 and 5 [3]. This in turn causes activation of neurons expressing pro-opiomelanocortin (POMC)/cocaine- and amphetamine-regulated transcript (CART) causing upregulation of α-melanocyte-stimulating hormone (α-MSH) [3]. The latter activates the melanocortin 4 (MC4) receptor located in the paraventricular nucleus of the hypothalamus and lateral hypothalamic area, leading to suppression of appetite and weight loss [3]. Leptin signalling causes suppression of orexigenic peptides, i.e. agouti-related peptide (AgRP) and neuropeptide Y (NPY) [3].
LepR deficiency is quite rare, first described in db/db mice [4, 5]. It is inherited in an autosomal recessive (AR) manner. Most of the cases are reported in families with a high degree of consanguinity. Individuals affected by LepR deficiency exhibit normal birth weight with rapid weight gain in infancy leading to early-onset obesity, hyperphagia, and impaired satiety. They develop a number of metabolic and hormonal abnormalities. Hyperinsulinaemia, early-onset diabetes, non-alcoholic steatohepatitis (NASH), and premature coronary artery disease (CAD) are some of the metabolic consequences of early-onset obesity. Adequate leptin signalling is permissive for the onset of normal puberty and pubertal growth spurt [6]. Leptin also influences the secretion of growth hormone-releasing hormone (GHRH), thyroid-releasing hormone (TRH), and gonadotrophin-releasing hormone (GnRH) [6]. Patients develop growth hormone deficiency with stunted height, secondary hypothyroidism, and hypogonadotropic hypogonadism [6]. In addition, these patients are found to have lower CD4 T+ cell count and are predisposed to various infections, indicating the role of leptin in the modulation of the immune system [7].
In this case report, we have described a novel homozygous mutation in the LepR of an infant presenting with monogenic obesity.
Case report
A 12-month-old male infant from Northwestern India was referred to our endocrine clinic because of excessive weight gain and hyperphagia. His birth weight was 3 kg (–0.75 standard deviations score [SDS]). He was born of normal vaginal delivery. There was no history of gestational diabetes, hypertension, hypothyroidism, or more than recommended weight gain in the mother during pregnancy. There was no history of consanguinity between the parents or any of the family members until the third generation. There was a history of early-onset obesity in one of the maternal grand-aunts and in 3 maternal uncles.
From 2 months of age, the child exhibited intense hyperphagia and impaired satiety, demanding feeding every half an hour. He started gaining weight rapidly reaching 10.5 kg at 3 months (+9.4 SDS), 15.0 kg at 6 months (+12.6 SDS), and 21.3 kg at one year of age (+13.1 SDS). He was able to hold his neck at 2 months of age, sit without support at 4 months, and crawl at 9 months. Currently, he could stand with support but found it difficult without support due to his excess weight. There was no history of lethargy, constipation, dryness of skin, excessive hair growth, phallic enlargement, appearance of striae, or seizures.
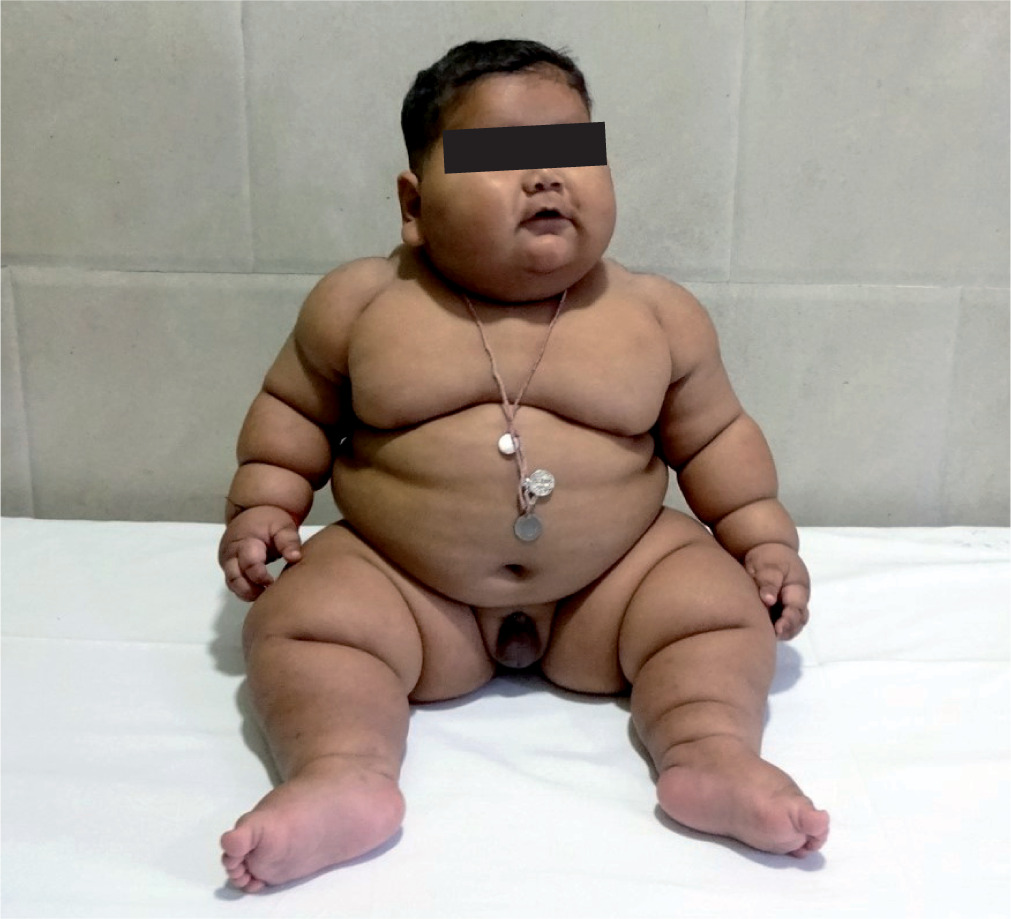
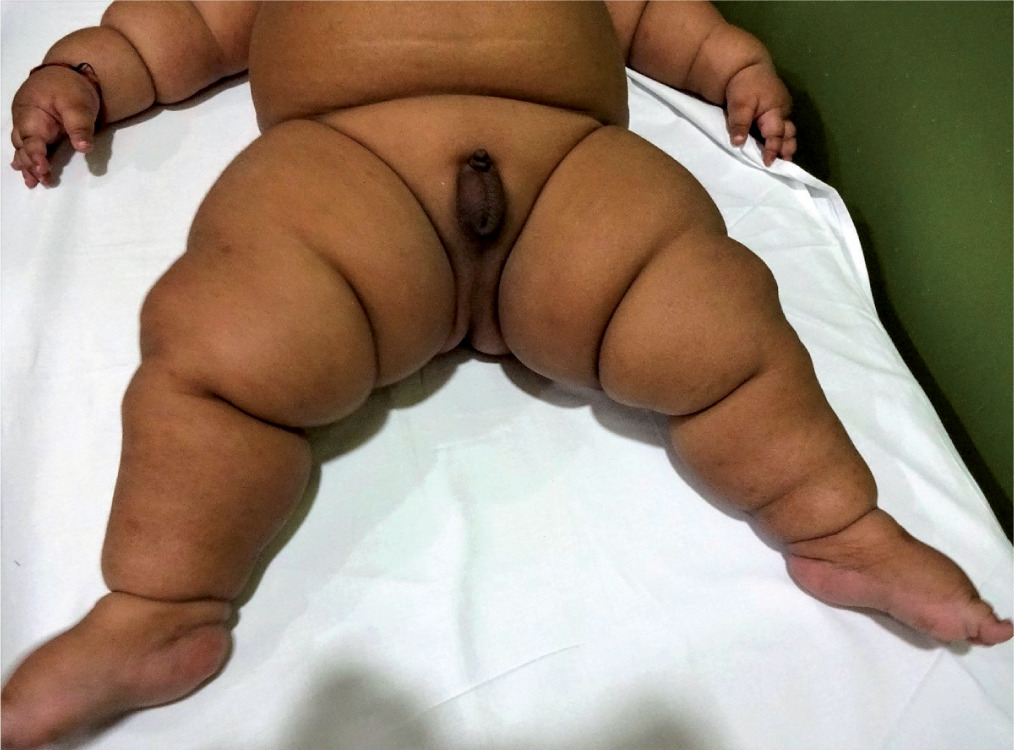
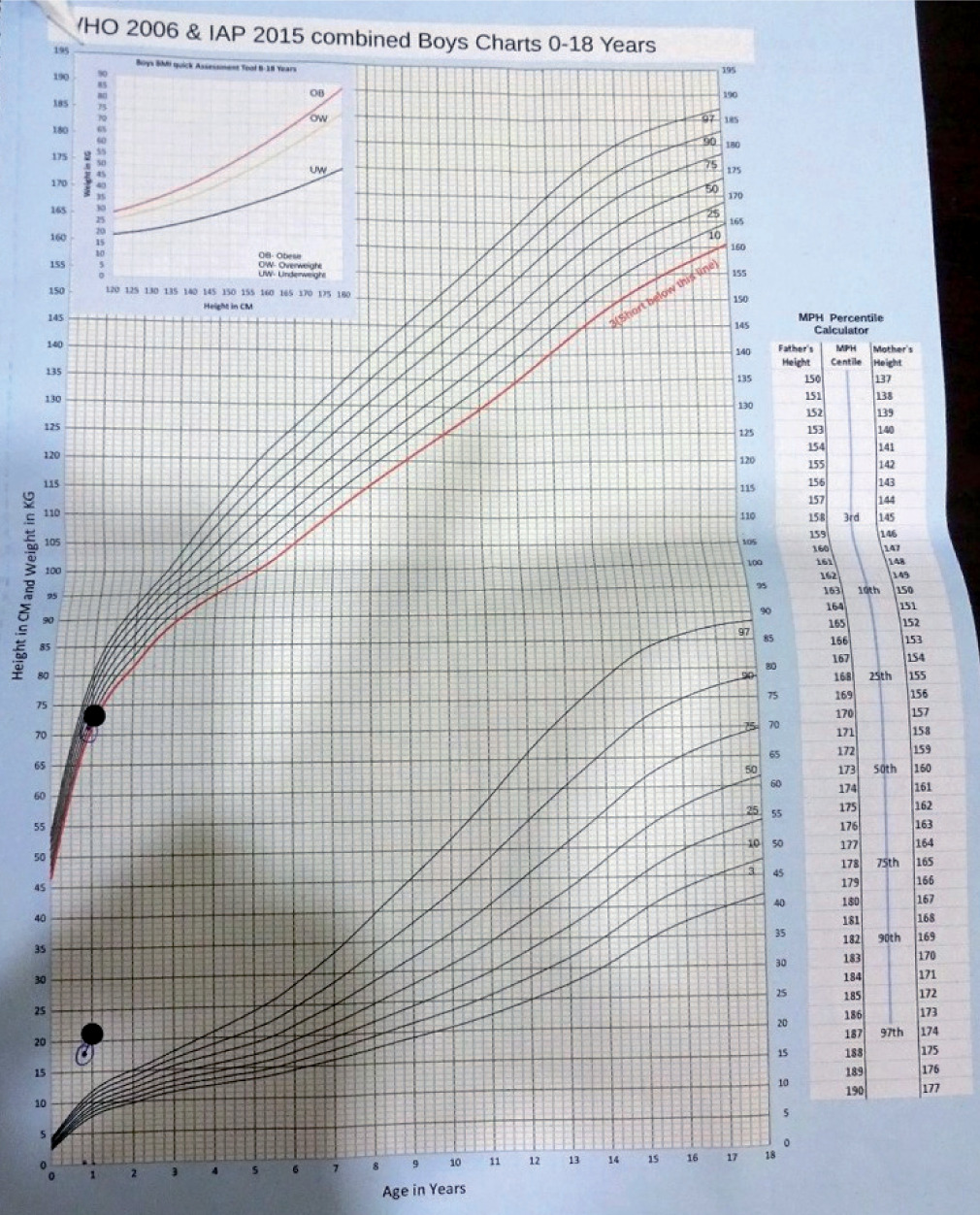
Physical examination revealed a generalized distribution of fat along with deep skin folds, predominant in the neck, abdomen, back, and upper and lower limbs (Figs. 1 and 2). Weight at one year of age was 21.3 kg (+13.1 SDS) and length was 73 cm (–0.68 SDS) (Fig. 3). Body mass index (BMI) was 40.2 kg/m2 (> 97th centile). The waist circumference measured midway between the lower ribs and the iliac crest was 50.5 cm (> 97th centile). His stretched penile length (SPL) was 2.8 cm, and pubic hair staging was P1. Central nervous system examination was normal, and there was no hypotonia of the muscles (unlike in Prader-Willi syndrome). There were no stigmata to suggest any syndromic disease, like any skin rash, short 4th metacarpal, or polydactyly. Eye examination was normal.
Figure 1
Clinical image of the patient showing early-onset obesity with a generalized fat distribution
Laboratory investigations revealed normal complete blood count, kidney function tests, normal fasting blood glucose, glycated haemoglobin, and fasting insulin level. However, liver enzymes were elevated, and the fasting lipid profile was deranged with elevated total cholesterol (TC), elevated triglyceride (TG), and low high-density lipoprotein (HDL). The hormone profile revealed normal thyroid function tests (thyroxine [T4] – 7.8 µg/dl [N 5.1–14.1 µg/dl], TSH – 1.73 uIU/ml, [N 0.7–6.4 normal uIU/ml]), ruling out primary hypothyroidism; normal serum cortisol (12.03 µg/dl [N 4.3–22.4 µg/dl]), and dehydroepiandrosterone sulphate (DHEAS) (30.8 µg/dl, N < 30 µg/dl), ruling out Cushing’s syndrome. The child also had normal serum calcium levels (9.3 mg/dl, N 8.5–10.5 mg/dl), ruling out the possibility of pseudohypoparathyroidism. However, serum leptin levels were markedly raised (71.92 ng/ml, N 2.05–5.63 ng/ml). The laboratory parameters are summarized in Table I.
Table I
Biochemical parameters of the patients
Suspecting monogenic obesity, genetic analysis was ordered after genetic counselling and obtaining informed consent from the parents. Genomic deoxyribonucleic acid (DNA) was extracted from the blood of the patient and target gene capture was performed using a custom capture kit. The constructed DNA libraries were sequenced to a mean depth of > 80–100X on the Illumina sequencing platform. Germline variants were identified using Genome Analysis Tool Kit (GATK) best practices framework. Gene annotation of the variants was performed using Variant Effect Predictor (VEP) programme [8] against the Ensembl release 99 human gene model [9]. Clinically relevant mutations in both coding and non-coding regions were annotated using published variants in the literature and a set of disease databases: ClinVar, OMIM (Online Mendelian Inheritance in Man), HGMD (Human Gene Mutation Database), LOVD (Leiden Open Variation Database), DECIPHER (Database for the Interpretation of Phenotype-linked Plausibly Pathogenic Sequence and Copy-number variation; population CNV), and SwissVar [10–15]. Common variants were filtered based on allele frequency in 1000Genome Phase 3, gnomAD (v3.1 & 2.1.1), dbSNP (GCF_000001405.38), 1000 Japanese Genome, TOPMed (Freeze_8), Genome Asia, and Medgenome’s internal Indian population database (MedVarDb v2.1) [16–21].
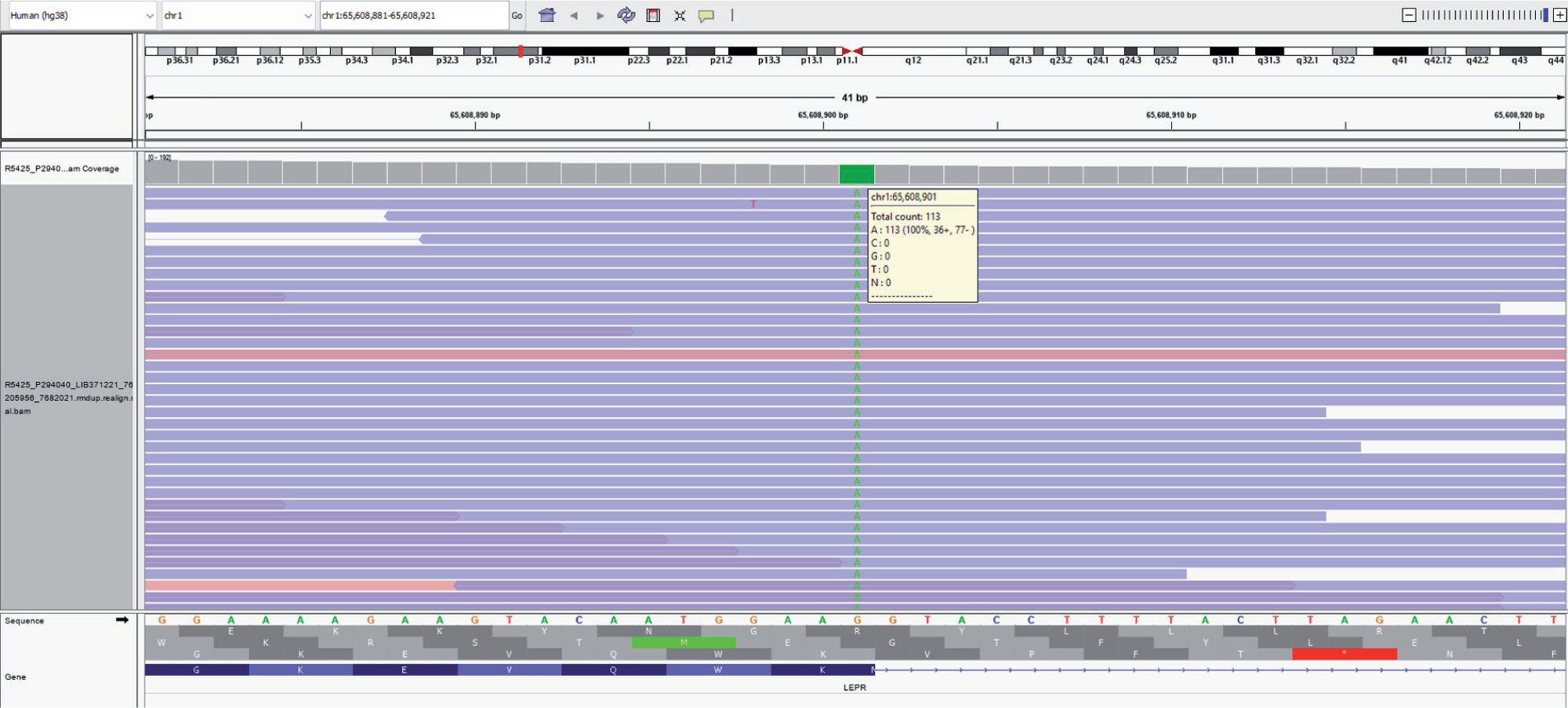
Targeted re-sequencing of relevant genes previously implicated in monogenic obesity revealed a novel homozygous mutation in exon 12 of the LEPR gene (chr1:g.65608901G>A) (Fig. 4). There was a change of a single nucleotide base from guanine to adenine that resulted in the synonymous amino acid change of Lysine at codon 584 proximal to the donor splice site (p.Lys584). This variant has not been reported in the 1000 genomes and genome aggregation database (genomAD). It was found to have a minor allele frequency of 0.002% in the internal database of MedGenome. The in silico prediction of the variant was “damaging” by MutationTaster 2. The reference codon was conserved across species. Due to a lack of evidence in the literature, the LEPR variation was classified as a ‘variant of uncertain significance’ and had to be correlated carefully with the clinical symptoms. It was recommended to perform Sanger sequencing validation of the specific variant identified from the next-generation sequencing (NGS) effort in parents and other family members to understand the segregation pattern of alleles. However, due to financial constraints the family could not afford the same at the time of writing this report.
Figure 4
Sequence chromatogram showing mutation in the exon 12 of LEPR gene (chr1:g.65608901G>A) resulting in the synonymous amino acid change of lysine at codon 584 proximal to donor splice site (p.Lys584)
The child was started on a supervised regimen of a low-calorie, low-carbohydrate, low-fat diet. Meanwhile, funds were being arranged to procure setmelanotide, an MC4 receptor agonist, found to be effective in LepR mutation.
Discussion
LepR deficiency is a rare endocrine disease. Kleinendorst et al. performed a comprehensive epidemiological analysis and systemic literature review to estimate the prevalence of LepR deficiency in Europe [22]. They found that, worldwide, 86 cases had been described in the literature, and they added 2 new patients, bringing the number of published cases to 88. This corresponded to a prevalence of 1.34 per million population. All patients had early-onset obesity, 96% had hyperphagia, and 34% had one or more pituitary hormone deficiencies [22].
Because the disease is autosomal recessive (AR) in the inheritance pattern, most of the cases are reported from families with a high degree of consanguinity. Most of the cases in the published literature are from the Punjab province of Pakistan [22]. Interestingly, our patient also hails from a geographical area along the border of the Indian Punjab province. Although there was no history of consanguinity between the parents or other family members, the geographical location points towards the process of natural selection or random genetic drift for the LEPR gene in this population [23].
To the best of our knowledge, there is only one previously reported case of LepR mutation from India. It was from a consanguineous family of 4 siblings, with one unaffected and 3 affected members. The family is from Western India [24]. Whole exome sequencing revealed frameshift mutation in the LEPR gene resulting in truncated LepR.
Early-onset obesity warrants genetic evaluation for monogenic obesity and syndromic causes. Monogenic obesity can be caused by mutations in the genes coding for leptin, LepR, MC4, prohormone convertase/subtilisin kexin 1 (PCSK1), and POMC, which are involved in the regulation of appetite and satiety [1]. Others include inactivating mutations of FTO, TUB, and FAT genes or overexpression of genes encoding for AgRP or NPY. Rare syndromic forms include Prader-Willi, Bardet Biedl, Alström, Carpenter, Cohen, and Beckwith-Wiedemann syndromes [1].
Serum leptin levels have been found to correlate positively with the amount of body fat and BMI in non-genetic/sporadic obesity [25]. Previous studies have shown that the serum leptin concentration cannot definitively distinguish between sporadic and monogenic obesity [26]. Also, because the prevalence of LepR mutation is quite low (which prevents the study of larger case series), there has been difficulty in establishing cut-offs of serum leptin for defining leptin resistance [26]. Thus, it is mostly the ‘clinical suspicion’ that should drive a physician to decide on the screening for LepR mutation.
The genetic mutation described in our patient (chr1:g.65608901G>A) has not been reported previously. The nucleotide base change from guanine to adenine occurred at codon 584 proximal to the donor splice site, resulting in a synonymous amino acid change of lysine (p.Lys584) at exon 12 of chromosome 1 containing the LEPR gene. A splice site mutation refers to an alteration in the DNA sequence occurring at the boundary of an exon (coding) and an intron (non-coding) [27]. Considering its proximity to the donor splice site and conservation of the reference codon across the species, it might affect the correct splicing and expression of the LEPR gene. One splice site mutation (G>A) resulting in skipping of exon 16, and truncated protein of LEPR, has been previously reported in a consanguineous family from Kabylian (Berber of Northern Algeria) in which 3 of 9 siblings had morbid obesity [28].
The functionality of the mutation was tested by in silico prediction tools using MutationTaster 2. It is known that these tools are poor in specificity, leading to a high false positive rate [29]. However, the in silico prediction correlates with the clinical and biochemical profile of the patient. For further confirmation, it is prudent to offer genetic counselling to all family members and do a genetic analysis of parents and others affected by obesity.
Recombinant leptin therapy would not be helpful in LepR mutation. The European Medicines Agency (EMA) has now recommended the use of a novel agent, setmelanotide (Imcivree, Rhythm Pharmaceuticals), for the treatment of POMC, PCSK1, LepR mutations, and Bardet Biedl syndrome [30]. In the USA, setmelanotide has received approval for chronic weight management in patients 6 years of age and above with obesity caused by POMC, PCSK1, and LEPR deficiency [30]. Setmelanotide binds to and activates MC4 receptors, thereby suppressing appetite and causing weight loss. The common side effects are injection site reactions, skin hyperpigmentation, and gastrointestinal side effects like nausea, abdominal bloating, and pain [31]. At the time of writing this report, we were trying to arrange funds to procure setmelanotide for our patient. It would be interesting to observe how the patient’s weight and hunger respond to the treatment. Meanwhile, the child was started on a supervised calorie-restricted diet. The family was also counselled to engage the child in physical activity for at least 20 minutes daily when he would be able to walk. Drugs like GLP-1 analogues (liraglutide in a dose of 3 mg) have been tried in monogenic obesity due to MC4 mutations, but without effective results [32]. Also, data regarding bariatric surgery in monogenic obesity have not been encouraging. In a small survey of 6 patients with LepR deficiency, who underwent bariatric surgery, 3 were not able to maintain the weight loss induced by surgery [33].
Conclusion
To conclude, we have described the case of a 12-month-old male infant who presented to us with severe obesity and hyperphagia and was found to have homozygous LepR mutation. The mutation described was a novel one and involved the synonymous amino acid change of lysine at codon 584 proximal to the donor splice site (p.Lys584). To the best of our knowledge, this is the second case report of LepR mutation from India. Identification of LepR mutation is important because setmelanotide can offer substantial benefits in these patients.









