
Introduction
The 22q11.2 deletion syndrome, known as DiGeorge syndrome, is caused by a microdeletion of chromosome 22, has an approximate occurrence of 1 in 4500 live births and it is the commonest interstitial deletion [1]. The phenotypic features of DiGeorge syndrome demonstrate wide variability and can present at any age. Most patients develop congenital cardiovascular anomalies (74% of patients), craniofacial anomalies (most patients), palatal anomalies (69%), immunodeficiency due to hypoplasia/aplasia of the thymus (77%), developmental delay or learning disabilities (70%–90%) and hypocalcemia associated with hypoparathyroidism (50%) [1]. Typically, the disease is diagnosed in early childhood, but with such a broad variation of signs and symptoms, DiGeorge syndrome is still often underdiagnosed. Sometimes the lack of characteristic symptoms, such as diagnosed hypocalcemic tetany, hemodynamically significant heart defect or obvious cleft palate, may lead to its diagnosis in the teenage years or even in adulthood, as in the case of our patient. In the current paper we present a case of a 13-year-old male child who presented with high creatine kinase level, which was found accidentally during his stay at the emergency department due to symptoms of gastroenteritis. This result led him, through neurologist and geneticist, to have an array-based comparative genomic hybridization examination. The test was made because of muscular dystrophy suspicion. The outcome was unexpected as tests for myodystrophy turned out to be negative, but simultaneously the diagnosis of DiGeorge syndrome was established. This is one of many cases of patients who were diagnosed in their teenage or adult years due to mildness and non-specificity of the symptoms [1–4]. According to the patient’s past medical history, there were many signs and symptoms that could have led to an earlier testing, but they were mild and unclear. The patient and his legal guardians have consented to the publication of the case report.
Case presentation
A 13-year-old male patient presented to the emergency department (ED) due to the symptoms of gastroenteritis. The boy was born by vaginal delivery after an uneventful pregnancy at the 39th week of gestation with a birthweight of 3520 g and Apgar score of 10. His neuromotor development was insignificantly delayed, with particular delays in the emergence of language. He started walking at 15 months and spoke his first words at the age of 10 months with subsequent speech delay at 18 months. In his early years, he underwent frequent urinary and respiratory tract infections and was hospitalized due to those conditions several times. He was also diagnosed with right aortic arch, submucosal cleft palate, mild impairment, requiring early development support, and speech impediment. He was taken care of by a cardiologist, laryngologist, neurologist, speech therapist and physiotherapist (Figure 1). At admission, his complaints were nausea and abdominal pain. In examination his vital signs were normal. He presented typical dehydration features such as dry oral mucosa and sunken eyes. He also had the following typical features of craniofacial anomalies: protruding, low-set auricles bilaterally, antimongoloid position of the eyes, long nasal dorsum and short philtrum. Laboratory studies performed at ED demonstrated ionized calcium level of 0.68 mmol/l (reference range 1.15–1.29 mmol/l) and increased creatine kinase (3059 IU/l; reference range 0–270 UI/l). Electrocardiography detected long QT. At the department he was treated symptomatically due to dehydration and abdominal pain and was referred to specialist clinics (cardiology, neurology, geneticist and gastroenterologist) with the suspicion of muscular dystrophy. After a few months, genetic tests were performed. Array comparative genomic hybridization (aCGH) showed a deletion of region q11.2 on one chromosome 22, consistent with DiGeorge syndrome. In the meantime, the patient underwent 2 afebrile seizure episodes lasting from few seconds to few minutes presented as hands contorting, loss of consciousness and upper lip elevation. A computed tomography exam was performed and revealed Fahr syndrome, a bilateral striatopallidodentate calcinosis. According to those seizures, after receiving the result of genetic examination, the patient was directed to the pediatric endocrinology clinic for further survey and treatment. At admission he was 168 cm tall (75th–90th percentile for sex and age according to Polish growth chart) and weighted 63.1 kg (75th–90th percentile), BMI 22.4 kg/m2 (90th percentile) (Figures 2, 3) [5]. At the department laboratory tests were performed (Table I). Blood morphology analysis did not show any significant abnormalities, renal function tests were normal and only alanine transaminase was slightly elevated. Thyroid function tests, including TSH, FT3 and FT4 levels, were within normal range. The relevant observations were hypocalcemia (1.53 mmol/l; reference range: 2.2–2.65 mmol/l), hyperphosphatemia (3.35 mmol/l; reference range: 1.09–2 mmol/l), parathormone level below the normal range (3.15 pg/ml; reference range: 15–65 pg/ml) and vitamin D deficiency. Creatinine kinase activity was still high (2399 U/l). In the daily 24-hour urine collection fractional tubular reabsorption of phosphate (TRP) was 98%. Another important clinical finding was hypercholesterolemia with high low density cholesterol (LDL-C) and low high density cholesterol (HDL-C) levels. Additionally, computed tomography (CT) was performed. It revealed multiple calcifications located bilaterally in the basal ganglia, dentate nuclei and frontal lobe. Another finding was septum pellucidum cyst. During EEG monitoring, mild seizure episodes were observed. During hospitalization, the appropriate treatment was implemented. The patient received 1 µg of alfacalcidol twice daily, 1000 mg of calcium three times a day, 470 mg of magnesium lactate three times a day and 1000 IU of vitamin D once a day. After almost two weeks of treatment normalization of calcium and phosphorus levels was observed. The patient was discharged home with recommendation for further multidisciplinary care. In addition to pharmacological treatment, a low-phosphorus diet was recommended.
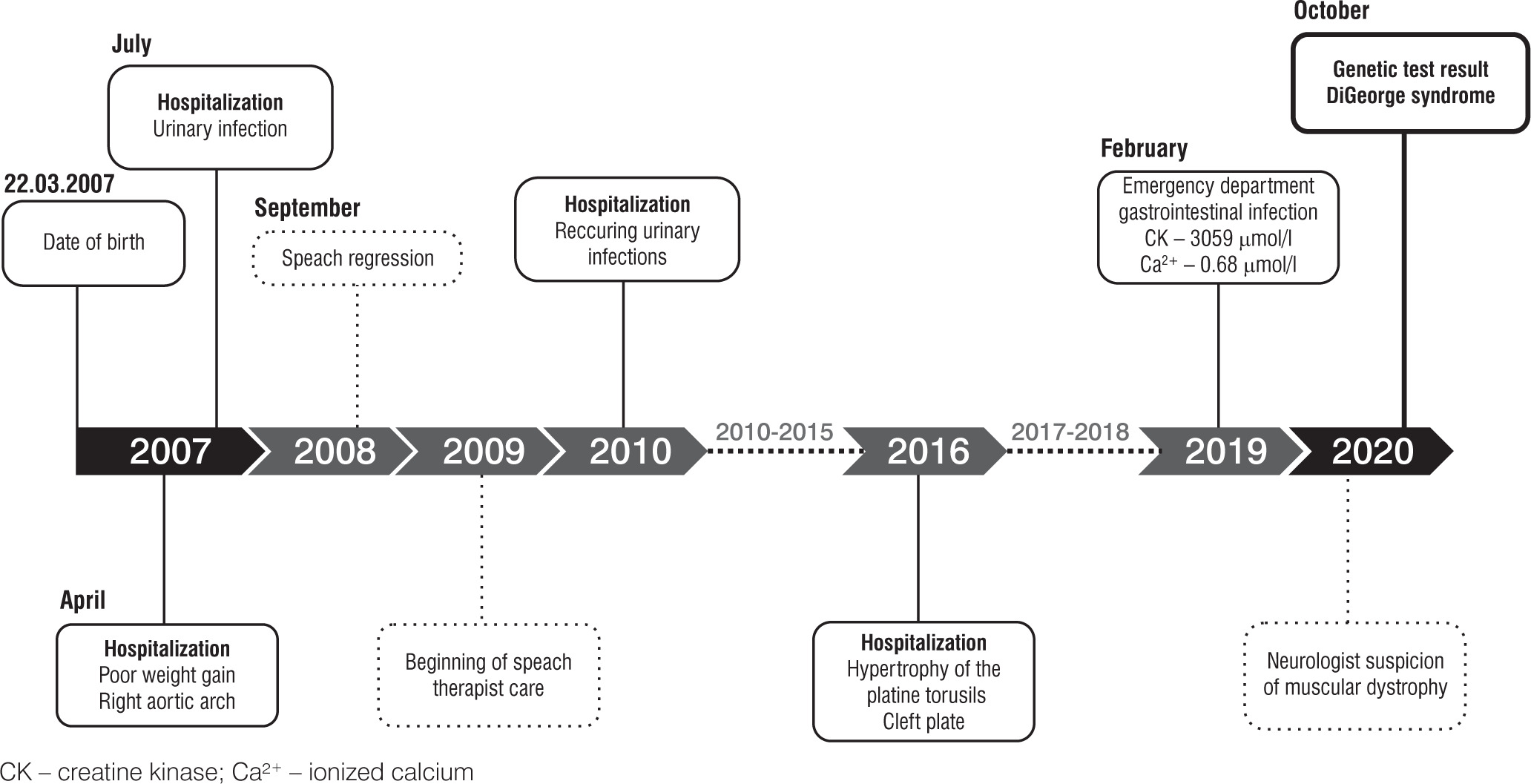


Table I
Laboratory results of the patient
Follow-up
After more than one year the patient reported to the pediatric department for medical supervision. At admission his general condition was good, with no disturbing symptoms. He was 174 cm tall (50th–75th percentile) and weighted 70.5 kg (75th–90th percentile), BMI 23.28 kg/m2 (75th–90th percentile) (Figures 2, 3) [5]. Chvostek’s and Trousseau’s signs were absent. No seizure episodes were observed since the last hospitalization. The biochemical markers of calcium-phosphate metabolism were within the reference range. Creatine kinase activity was normal. Cholesterol level was still elevated and vitamin D concentration was suboptimal. Dual-energy X-ray absorptiometry (densitometry; DXA) did not show decreased bone density; Z-score for lumbar spine (LS) measurement was 1.6 and the total body less head (TBLH) was 0.4. Daily 24-hour urine collection was performed twice. Both examinations showed increased calcium excretion level with fractional tubular reabsorption of phosphate (TRP) within normal range. According to the patient’s condition, blood tests and imaging techniques surveys, previous treatment was successful and well tolerated by the patient. The decision of medication maintenance was made.
Discussion
The presented case is a description of the patient who was diagnosed with DiGeorge syndrome at the age of 13. Typically, the disease is diagnosed in early childhood, but sometimes the lack of characteristic symptoms, such as diagnosed hypocalcemic tetany, hemodynamically significant heart disease or obvious cleft palate, may lead to the diagnosis in the teenage years or even in adults, as in the case of our patient. The average age of a diagnosis varies from 0 to 33 years and depends mostly on the presented symptoms [6, 7].
In the boy, the syndrome was manifested by subclinical abnormalities that did not arouse the doctors’ suspicion of DiGeorge syndrome. The child had: right-sided aortic arch, submucosal cleft palate, non-evident hypocalcemia signs (tremor) and non-specific, slight features of facial dysmorphism. Hypocalcemia is observed in about 70% of patients with the 22q11.2 mutation, but its symptoms are not always observed. With latent hypoparathyroidism, PTH (parathyroid hormone) is secreted in quantities that are enough to maintain normal calcium and phosphorus concentrations, however, during the period of increased demand for calcium, such as during puberty, PTH secretion becomes insufficient, which contributes to the symptomatic manifestation of hypocalcemia [8]. On the other hand, hypocalcemia related to hypoparathyroidism is not presented in every patient, may develop later, be persistent or transient and is often masked by the other signs or symptoms. Even if hypocalcemia is present, patients are often well adapted to long lasting low calcium levels and can develop symptoms while its concentration is extremely low.
In the presented patient, high creatine kinase activity raised the suspicion of myopathy. For this reason a genetic test was performed, which showed the presence of the 22q11.2 mutation, without previous suspicion of DiGeorge syndrome. The literature also presents cases of patients of different ages with an atypical manifestation of undiagnosed DiGeorge syndrome. For example, in an almost 12-year-old boy, episodes of hypocalcemia manifested in the form of laryngospasm and tonic seizures [8]. In another publication Johnston et al. presented the case of a 29-year-old woman who saw a general practitioner because of recurrent numbness and tingling of the hands and feet for 2 years. In this case a transient hypocalcemia was detected in the neonatal period, which resolved after appropriate calcium and vitamin D supplementation. In subsequent years no hypocalcemic episodes occurred and the patient underwent a series of tests due to numbness, including genetic testing, then the 22q11.2 mutation was detected [9]. There are also publications of cases in which hypocalcemia did not give any overt symptoms and was detected accidentally. This happened in the case of 11-year-old Korean girl. She was reported to emergency department due to the fever and vomiting. Laboratory tests revealed hypocalcemia and decreased parathyroid hormone level. Later a microdeletion on chromosome 22q11.2 was confirmed by fluorescence in situ hybridization. Looking at her past medical history, she was born with imperforate anus with rectovestibular fistula and partial cleft palate, but there was no history of neonatal hypocalcemia and karyotyping by GTG banding was normal. In the following years, she had frequent respiratory tract infections, chronic constipation, development delays, mild intellectual disability and mildly dysmorphic facial features, but due to the lack of typical symptoms, the DiGeorge syndrome was not suspected [10]. Diagnose of DiGeorge syndrome is not simple even in patients with documented hypocalcemia, as it was in the case of 44-year-old woman who suffered from recurrent syncope episodes. She also had QT-prolongation due to hypocalcemia. As a child she experienced epileptic seizures, but no further treatment was required. Only after finding not previously documented mild facial dysmorphia, she was directed for genetic testing, which revealed a microdeletion in 22q11.2, confirming the diagnosis of DiGeorge syndrome [11].
Psychiatric symptoms may also be a manifestation of this disease. Patients with DiGeorge syndrome have a risk of psychiatric disorders up to three times higher than a healthy population, which was shown in a Danish study published in 2017 [12]. There are many case reports of genetically undiagnosed patients with 22q11.2 deletion syndrome presenting psychiatric disorders. Interestingly, there might be a lot of patients over 40 year old, who remain undiagnosed, due to the fact that the genetic testing was not available before the nineties of the twentieth century [13].
Rizvi et al. described the case of a 21-year-old African-American woman in whom DiGeorge syndrome manifested in the form of psychosis. A CT scan of the head showed calcification of the basal ganglia, a Fahr syndrome was also detected in the patient and laboratory tests revealed hypocalcemia and hyperphosphatemia [14]. In other patient, the 51-year-old man, the DiGeorge syndrome was diagnosed after many years of treatment due to the panic disorder and a severe depressive episode with suicidal ideation. As a child, this man underwent a heart surgery for septal defect at 6 years of age, correction of polydactyly (two additional thumbs) at 6 years of age, and polypectomy in the nose (10 years of age) and then the colon (51 years of age). Geneticist was consulted because all of the symptoms above, but only after severe psychiatric episode occurred [13].
The high activity of creatine kinase in our patient was present due to previously undiagnosed tremor, without obvious exponents of hypocalcemic tetany, but leading to muscular cell destruction with a release of rhabdomyolysis markers, as well as leakage of the enzyme from the myocytes due to the membrane permeability under the influence of hypocalcemia [15–17]. In the patient myopathy was suspected because of high activity of creatine kinase. Low levels of calcium in the blood were accidentally detected several times, however it was not noticed by doctors and the diagnosis was not extended. This patient is not the first described case in whom hypoparathyroidism has been mistaken for muscular disease. In the literature, there are similar cases of patients whose muscle damage due to hypocalcemia was previously misdiagnosed as polymyositis or myopathy [18–22]. Combination of these conditions indicates the need for a more in-depth analysis of laboratory tests and differential diagnosis, as the treatment of hypoparathyroidism is quite simple and the patient’s condition improves in a short time after initiation of appropriate treatment [20]. In the patient with hypothyroidism-induced myopathy due to hypocalcemia, after correction of hypocalcemia with 600 mg calcium carbonate twice daily and 0.75 µg of calcitriol daily, creatine kinase levels gradually normalized [20]. However, it is crucial that in the patients with a parathyroid hormone deficiency, only active forms of vitamin D should be used, because the hydroxylation of 25-hydroxyvitamin D to 1.25-hydroxyvitamin D is a process that occurs with the participation of PTH and its deficiency disturbs this reaction [23, 24].
Conclusions
DiGeorge syndrome can sometimes cause non-specific, subclinical symptoms that do not at first glance indicate the presence of the disease, but doctors should always be careful in the presence of hypocalcemia of unknown origin and conduct a thorough clinical examination, medical interview, and analyze the patient’s medical records. There is a need to determine the level of calcium and phosphate levels in patients with non-specific muscular symptoms and with high activity of creatine kinase. The correct interpretation of these laboratory results may significantly shorten the procedure of diagnosis and make the correct reconnoitring.









