
Introduction
Primary hyperparathyroidism (PHPT) is a rare disease in children and adolescents. It is more commonly seen in post-menopausal females [1]. In Western countries, it is mostly diagnosed incidentally during routine biochemical screening [1]. The classical manifestations of PHPT, i.e. stones, bones, groans, psychic moans and fatigue overtones, are rarely seen in the developed world. However, in countries like India, the disease still continues to manifest as a florid skeletal and renal disease [2].
The incidence of PHPT is very low in children, at 1/300,000 live births/year, and the prevalence is 3–5/100,000 [3].
Pediatric PHPT differs from the adult variant in multiple aspects. In children and adolescents, the disease may have a myriad of manifestations, ranging from asymptomatic to florid course. The disease may be associated with multi-glandular and more aggressive histopathology of parathyroid tumours. It can also be a presentation of various syndromic disorders like multiple endocrine neoplasia type 1 (MEN1), multiple endocrine neoplasia type 2A (MEN2A), multiple endocrine neoplasia type 4 (MEN4), hyperparathyroid jaw tumour.
The literature on pediatric PHPT is so far limited to a few case reports and case series [4–9].
The single centre experiences have mostly reported retrospective databases [10–16]. There is a paucity of knowledge regarding the long term outcomes, recovery, recurrence of the disease and development of syndromic features.
In the present study, we report our experience with 10 cases of pediatric PHPT, their spectrum of manifestations and their follow-up after 6 months and 1 year of curative surgery. Our study highlights the clinical, biochemical, radiological profiles of pediatric PHPT and also gives an insight into the trend of recovery after surgery.
Material and methods
We included 10 consecutive PHPT patients below 20 years of age attending the Endocrine clinic or admitted through the emergency services of our institute between March, 2018 and August, 2019. PHPT was diagnosed on the basis of PTH dependent hypercalcemia, i.e. serum total calcium of 2.6 mmol/l or higher (normal range: 2.1–2.6 mmol/l) and non-suppressed or inappropriately normal PTH levels (more than 2.1 pmol/l). Two of our patients had serum total calcium less than 2.6 mmol/l, with severe vitamin D deficiency [serum 25(OH) D less than 25 nmol/l] as defined by the Institute of Medicine [17]. However, PHPT was suspected in the setting of inappropriately elevated serum parathyroid hormone (PTH) and typical clinical and radiological manifestations.
After admission, we obtained their clinical data through a pre-designed proforma including age at presentation, symptoms, duration and presence of end organ damage like bone deformity, pancreatitis, renal stones, peptic ulcer disease. Physical examination was conducted to look for any bone deformity (like genu varum, genu valgum, windswept deformity, coxa vera, cubitus valgus), bone tenderness, spinal tenderness, abdominal tenderness. Family history was also enquired to distinguish any familial clustering of similar disease. The syndromic forms of PHPT were screened by looking for history of galactorrhoea, accelerated height spurt, neuroglycopenic symptoms, hyper-tension, adrenergic spells, goitre, and jaw tumour.
Blood samples were drawn in the fasting state. The serum total calcium, inorganic phosphorus and alkaline phosphatase were measured on Hitachi 917, Roche, Germany (normal range 2.1–2.6 mmol/l, 0.8–1.5 mmol/l and 80–240 IU/l, respectively). The intra-assay and inter-assay coefficients of variation were 3.5 to 5.0%. Serum inorganic phosphorus levels were interpreted based on their age matched cut offs.
Serum intact PTH (iPTH) was measured by chemiluminescence (Elecsys-2010, Roche, Germany; minimum detection limit = 0.1 pmol/l, normal range: 1.5–6.8 pmol/l). Urinary calcium, phosphate, creatinine were measured with automated analyzers. Hypercalciuria was defined as 24-hour urinary calcium more than 0.1 mmol/kg body weight/day (more than 4 mg/kg body weight/day) [18, 19]. Urinary calcium creatinine ratio (UCa/Creat) was also calculated and cut off for hypercalciuria was defined as more than 0.13 [20]. Serum 25 (OH) vitamin D was measured by chemiluminiscence (LAISON, DiaSorin, Inc., MN, normal range: 50–125 nmol/l) with coefficient of variation of 2.9–5.5%. Areal BMD was measured by dual energy X-ray absorptiometry (DXA), (Discovery A 84023, Hologic Inc., MA,USA) at lumbar spine (L1–L4, antero-posterior), left hip and non-dominant forearm as per the guidelines of the International Society for Clinical Densitometry (ISCD). Fractured vertebrae, vertebrae with prominent syndesmophytes, osteophytes, ligamentous calcification were excluded. BMD was recorded in terms of absolute mineral content in g/cm2 at various sites.
Localization of tumour was done with a functional scan using a Technetium 99 SESTAMIBI and a structural imaging, that is, 4 dimensional computed tomography (4D CT) of the neck. Pediatric PHPT is commonly associated with multiglandular disease and 4D CT neck has greater sensitivity than SESTAMIBI scan for detection of the same [21]. However, tumours at ectopic locations (like the mediastinum) could be adequately localized by Technetium SESTAMIBI scan. Hence, both the scans were used for localization of tumour, taking into consideration the possibility of multiglandular disease and ectopic parathyroid adenoma. After adequate localization, patients were subjected to parathyroidectomy.
Post operatively, patients were managed with oral calcium and vitamin D (cholecalciferol 60,000 units once weekly) supplements. Patients who had hungry bone syndrome were managed with alphacalcidol and intravenous calcium infusions.
Successful surgery was defined as normalization of calcium homeostasis, sustained over 6 months [22].
After surgery, patients were followed-up at 6 months and 1 year for re-assessment of clinical, biochemical and densito-metric parameters.
Statistical analysis
Statistical analysis was done using IBM SPSS Statistics© 23 (Armonk, New York, USA). Normative data are expressed as mean ± SD. Non normative data are expressed as median with interquartile range (IQR 25th–75th). Parametric tests (indepen-dent sample t test) were used to test significance between two independent means. Wilcoxon sign rank test and Mann-Whitney U test were used to test significance between non-parametric variables (paired and unpaired variables respectively). A value of p < 0.05 was considered significant.
Results
The 10 patients included 6 females and 4 males with a mean age of 16.7 ±1.8 years (age ranging from 14 to 19 years). The mean body mass index (BMI) was 18.4 ±3.9 kg/m2. The median duration of symptoms was 30 months (IQR 12.0– 51.0 months). The baseline characteristics of the individual patients are summarised in Table I. The clinical, radiological and histopathological results are summarised in Table II. The biochemistry results at baseline and after one year of surgery are summarised in Table III.
Table I
Baseline characteristics of individual patients
Table II
Baseline demographic, clinical, radiological parameters of the patients
Table III
Biochemical parameters of the patients at baseline, and 6 and 12 months after surgery
Spectrum of clinical manifestation
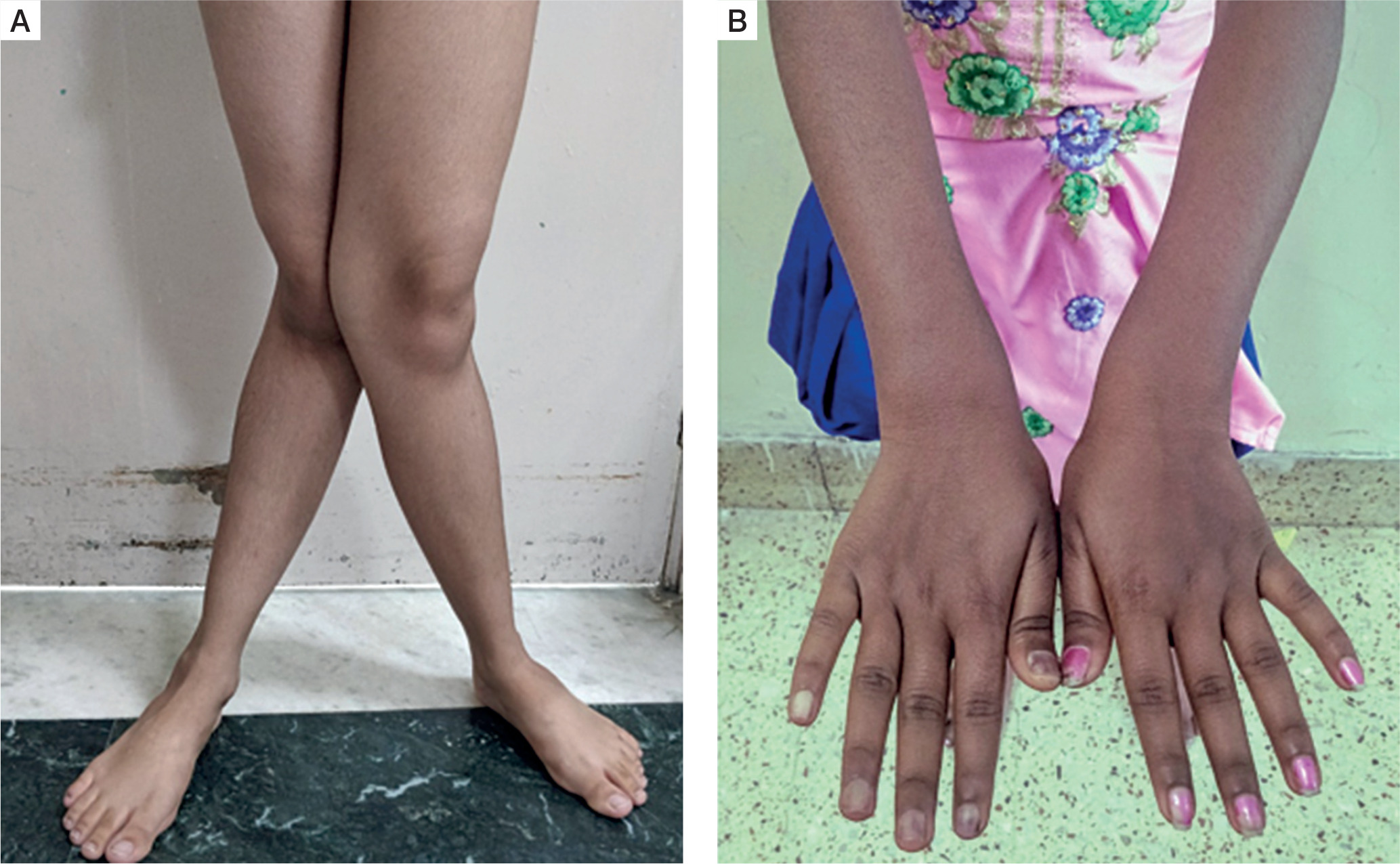
The predominant clinical presentation was musculoskeletal involvement in the form of musculoskeletal pain, described as diffuse nonspecific dull aching pain (90%). Bone deformity was present in 50% of the patients manifesting as various degrees of genu valgum (Fig. 1A). One patient had cubitus valgus in addition to genu valgum, while 4 patients had widening of wrists (Fig. 1B). None of the patients had genu varum, windswept deformity, rachitic rosary, or Harrison’s sulcus.
Figure 1
A) Severe degree of genu valgum seen in a female patient with PHPT. B) Widening of wrists seen in a female patient with PHPT
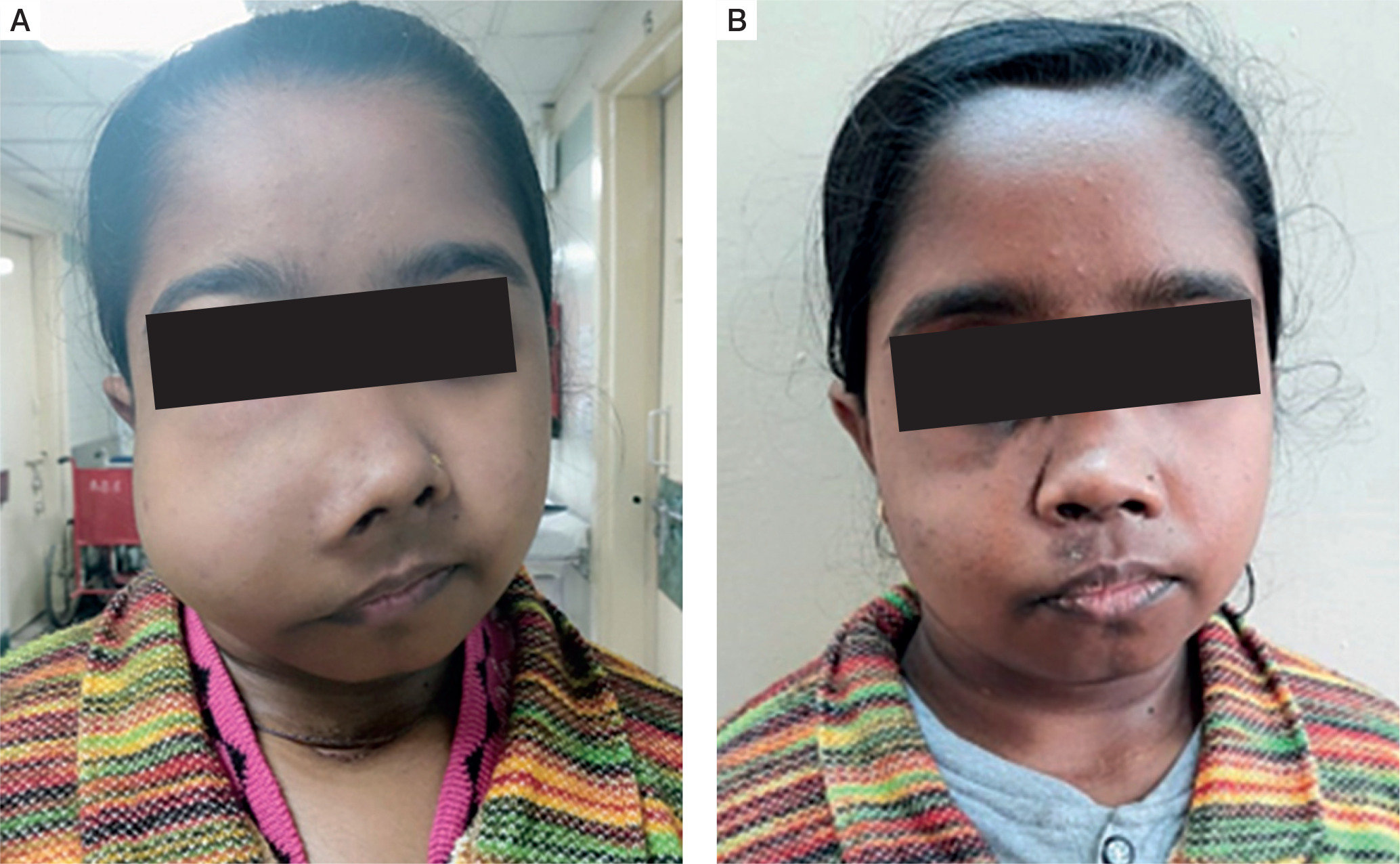
Three patients (30%) had history of fracture, with 2 patients completely bed ridden due to fractures of neck of femur. Four patients (40%) had proximal myopathy of lower limbs manifesting as difficulty in climbing upstairs or getting up from squatting position, which was confirmed on physical examination. Two patients had jaw tumours, one of which is shown in Fig. 2A, that caused facial disfiguration. She underwent surgery of her jaw tumour and facial reconstruction after 1 year of parathyroid surgery, as shown in Fig. 2B.
Figure 2
A) Jaw tumour seen in a patient with PHPT. B) Image of the same patient after removal of the jaw tumour
Four patients (40%) had symptoms related to the gastrointestinal system such as anorexia, severe reflux symptoms and constipation. Three patients (30%) presented with severe acute pancreatitis that was managed conservatively.
Five patients (50%) had renal stones detected on ultrasonography. None of the patients had neuropsychiatric manifestations in the form of depression, delirium, altered sensorium.
All the females in the cohort had attained menarche at the time of presentation. However, two out of 6 females had secondary amenorrhoea for more than 6 months.
Biochemical and radiological investigations
The mean serum total calcium was elevated, that was, 3.1 ±0.5 mmol/l (normal range: 2.1–2.6 mmol/l) with low normal serum inorganic phosphorus, 0.9 ±0.3 mmol/l (normal range: 0.8–1.5 mmol/l). Serum alkaline phosphatase (ALP) level was markedly elevated with median value of 1911.5 IU/l (IQR: 522.7–5702.3 IU/l; normal range: 80–240 IU/l).
Serum iPTH was significantly elevated with median of 133.5 pmol/l (IQR: 69.5–178.7 pmol/l; normal range: 1.5–6.8 pmol/l). Serum 25(OH)D level was low with a median value of 47.7 nmol/l (IQR 23.7–72.7 nmol/l; normal range: 75–250 nmol/l). Vitamin D deficiency was defined was defined as 25(OH)D level less than 50 nmol/l, while vitamin D insufficiency was defined as 25(OH)D level as 52.5–72.5 nmol/l, as per the Institute of Medicine guidelines [17]. Vitamin D sufficiency was defined as 25(OH)D level 75–250 nmol/l [17]. At presentation, 5 out of 10 patients (50%) had vitamin D deficiency, 3 patients (30%) had vitamin D insufficiency and 2 (20%) were vitamin D sufficient.
Urinary calcium was available for 9 out of 10 patients. Seven out of 9 patients (77.7%) had hypercalciuria, defined as 24 hour urinary calcium ≥ 0.1 mmol/kg body weight/day [18, 19]. Urinary calcium creatinine ratio was also calculated for 9 patients.
The median value for 24 hour urinary calcium was 10.5 mmol/day (IQR: 4.3–15.1 mmol/day, normal range: 2.5– 6.25 mmol/day). The mean urinary calcium/creatinine ratio at presentation was 0.5 ±0.2 (normal range, less than 0.13) [20].
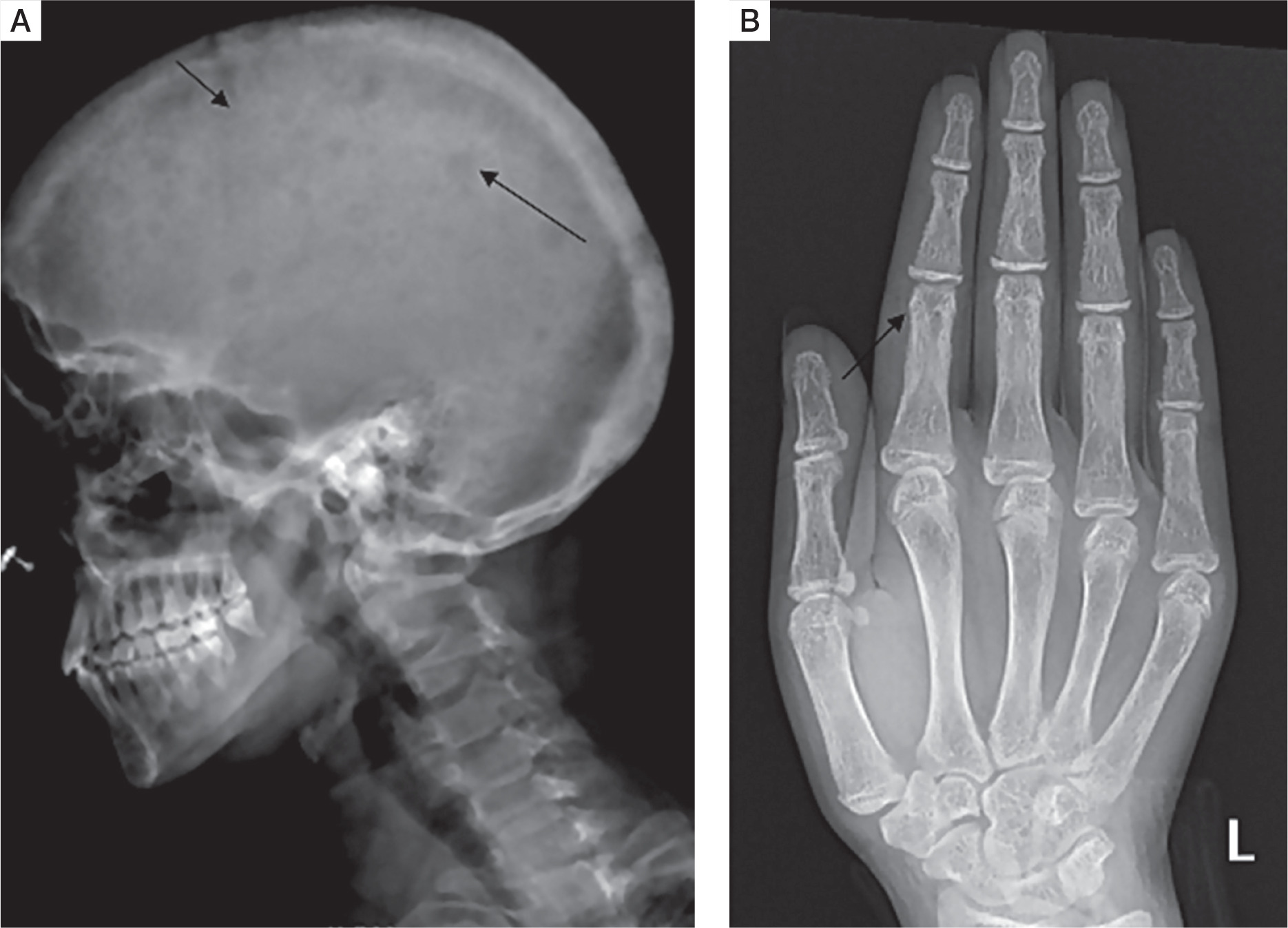
Skeletal survey was done in all patients. Osteitis fibrosa cystica was present in 5 out of 10 (50%), salt and pepper appearance of skull (Fig. 3A) in 5 (50%), subperiosteal resorption of phalanges (Fig. 3B) in 8 (80%), osteopenia in 9 (90%), pseudofracture in 6 (60%) and fracture neck of femur in 2 (20%) of patients. Pelvic radiographs of 4 patients showed tri-radiate pelvis, with osteopenia, subchondral resorption of sacroiliac joints and pubic symphysis (Fig 4A). Jaw radiographs of 5 patients showed loss of the lamina dura (Fig 4B), the thin lining of compact bone adjacent to the alveolus of tooth. Vertebral fracture (VF), detected on radiographic images of the spine (from T4–L4), was present in 4 out of 10 patients (40%), with 3 patients having more than one VF.
Figure 3
A) Lateral radiograph skull of a PHPT patient showing small well defined lucencies in calvaria giving a “pepperpot appearance”. B) PA view radiograph of left hand showing subperiosteal bone resorption, predominantly affecting the radial aspects of middle phalanges of second and third digits. Cortical tunneling is also noted
The mean bone mineral density (BMD) at lumbar spine (L1–L4) was 0.645 ±0.199 g/cm2, femoral neck (FN) 0.557 ±0.221 g/cm2, total hip (TH) 0.604 ±0.198 g/cm2 and distal forearm (DF) was 0.427 ±0.165 g/cm2.
Localization of parathyroid adenoma
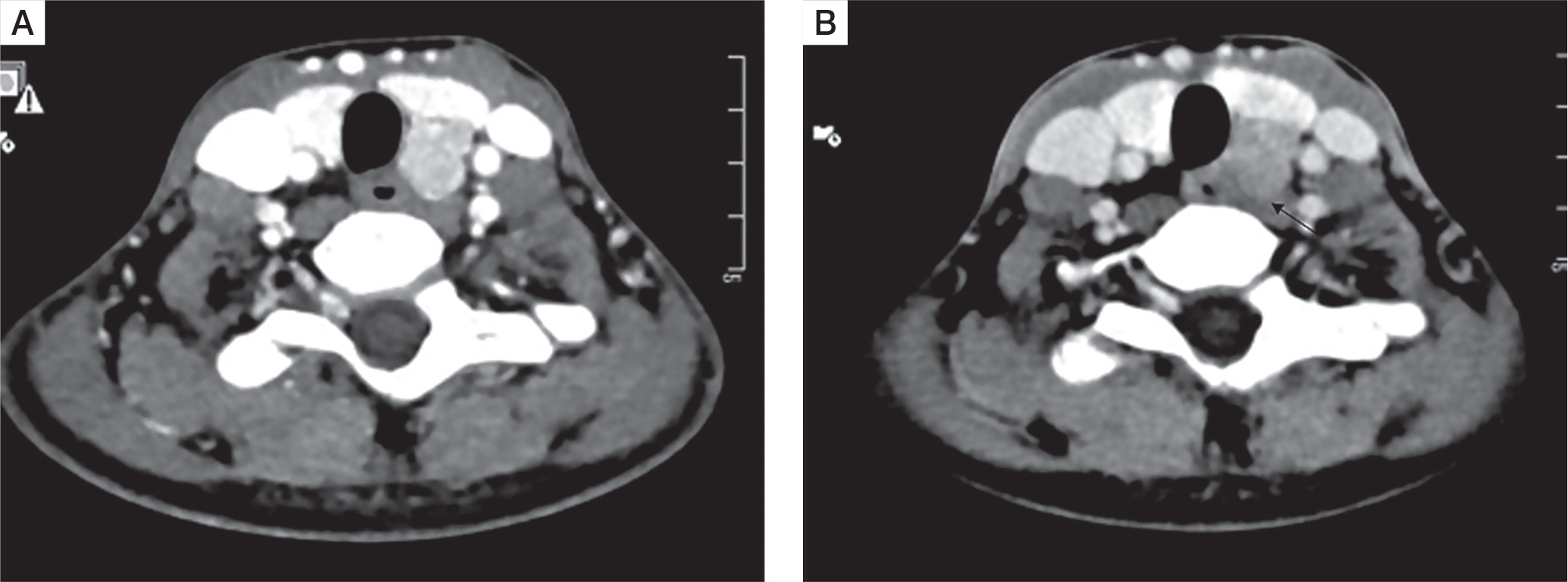
All patients had a single parathyroid adenoma localized on both Tc99 SESTAMIBI and 4D CT scans (Fig. 5A and B)]. Five out of 10 (50%) had left inferior parathyroid adenoma, followed by right inferior (3 patients), while one patient each had left superior and ectopic parathyroid adenoma in the anterior mediastinum. The mean size of parathyroid adenoma, as documented on 4D CT, was 1.8 ±0.7 cm.
Figure 4
A) Frontal radiograph pelvis of a PHPT patient showing generalized osteopenia, subchondral resorption of sacroiliac joints and pubic symphysis. B) Lateral radiograph of maxilla and mandible showing loss of lamina dura
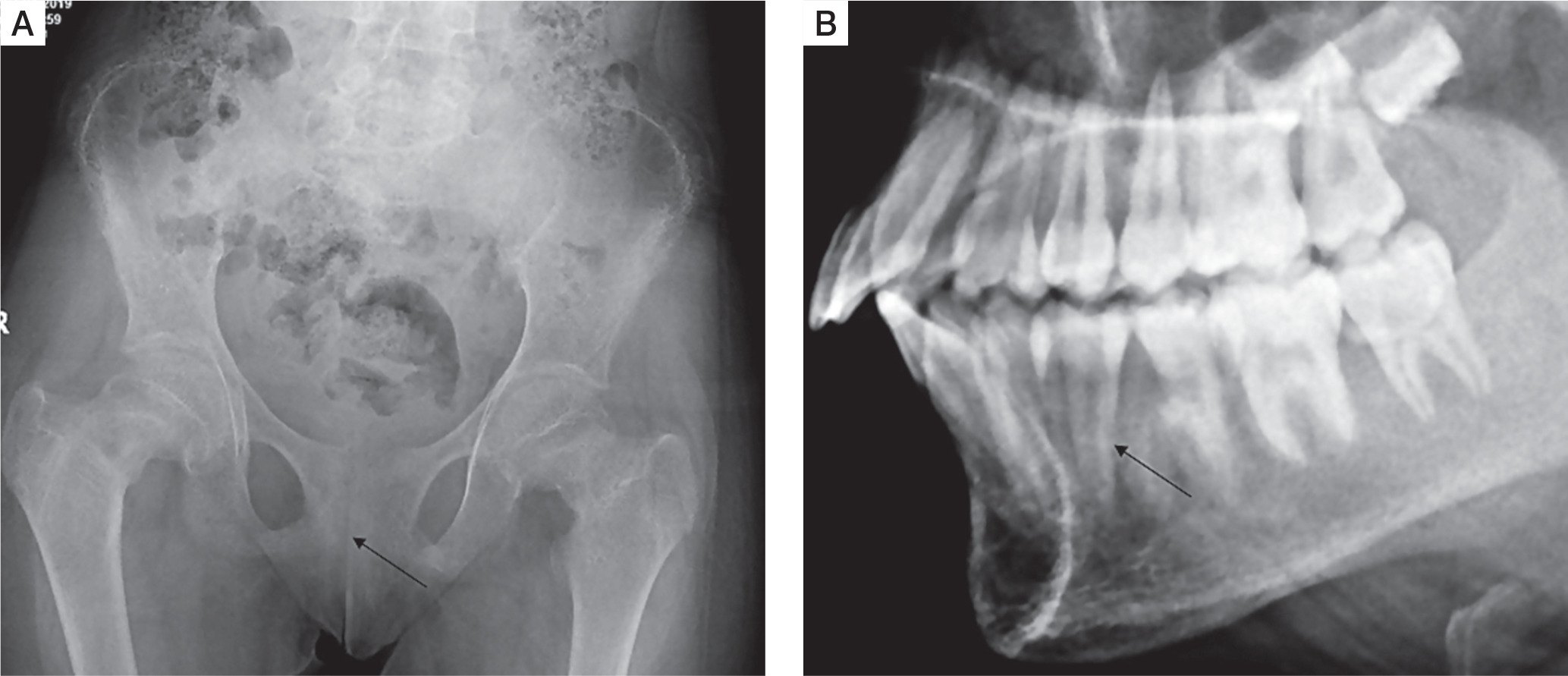
Figure 5
A) Axial computed tomography image (arterial phase) showing an oval hyperenhancing nodule with washout on delayed phase, in the region of left inferior parathyroid gland suggestive of left inferior parathyroid adenoma. B) Axial computed tomography image (arterial phase) showing washout on delayed phase suggestive of a parathyroid adenoma
Perioperative management
Pre-operatively, all patients with serum total calcium more than 2.7 mmol/l were managed with saline diuresis. In addition, 2 patients with serum calcium more than 3.5 mmol/l were administered Zoledronic acid 4 mg intravenous single dose, while one patient received Calcitonin 200 IU subcutaneous 6 hourly for five days. Zoledronic acid was administered 3 days prior to surgery. Both the patients developed fever and chills that lasted for only one day. At the time of surgery, the symptoms of hypercalcemic crisis had resolved and serum total calcium had fallen into the normal range of 2.1–2.6 mmol/l.
All patients were administered vitamin D supplementation 60,000 IU once weekly prior to surgery to prevent hungry bone syndrome.
All patients were subjected to focused parathyroidectomy by a single experienced surgeon. Single parathyroid adenoma was identified during resection in all cases. Histopathology revealed parathyroid adenoma with normal parathyroid tissue at the periphery in nine cases, while one patient had atypical adenoma with high mitotic activity and MIB-1 labelling index of 8%. The median tumour weight was 2.0 grams (IQR: 1.5–3.9).
Post-operatively, samples for serum calcium were sent everyday till one week. A decline in serum calcium was observed 48 hours after surgery. The mean post operative serum calcium after 48 hours was 2.1 ±0.4 mmol/l (normal range: 2.1–2.6 mmol/l). Immediate post operative serum iPTH observed after 24 hours was 7.6 pmol/l (normal range: 1.5–6.8 pmol/l). Symptomatic hypocalcemia manifesting as perioral tingling, numbness, paraesthesia was observed in 8 (80%) patients.
All patients were started on elemental calcium 1–2 g per day. Vitamin D 60,000 IU once weekly was continued in the post operative period. Patients with serum calcium less than 2.1 mmol/l were also initiated on alphacalcidol 1–2 µg per day.
Hungry bone syndrome defined as severe hypocalcemia (serum calcium ≤ 2.0 mmol/l, lasting beyond 96 hours of surgery), low phosphate, elevated ALP [23] was observed in 4 patients (40%). These 4 patients required intravenous calcium supplementation in addition to other aforementioned measures. Two out of these 4 patients continued to have severe clinical and biochemical hypocalcemia for a prolonged period and were discharged after 2 months of surgery.
Follow-up was available for 9 patients. At 6 months post-surgery, all patients stated significant improvement in symptoms of musculoskeletal pain, fatiguability, anorexia, abdominal pain. The mean serum total calcium after 6 months was 2.3 ±0.1 mmol/l (normal range: 2.1–2.6 mmol/l), mean serum inorganic phosphorus was 1.3 ±0.2 mmol/l (normal range: 0.8–1.5 mmol/l). The median values for serum ALP were 472.0 IU/l (IQR: 214.0–642.0 IU/l, normal range: 80–240 IU/l), serum intact PTH 8.3 pmol/l (IQR: 5.3–28.0 pmol/l, normal range: 1.5–6.8 pmol/l) and 25(OH)D 96.3 nmol/l (IQR: 67.5– 120.7, normal range: 75–250 nmol/l).
After one year of surgery, there was further improvement in clinical symptoms. All true and pseudofractures had healed. However, residual bone deformities had persisted, although there was no further progression. One patient with genu valgum whose ALP had normalized underwent surgical correction of genu valgum.
The patient with ossifying fibroma of the jaw underwent re-section of the tumour along with facial reconstruction.
The mean serum total calcium after one year was 2.3 ±0.2 mmol/l, mean serum inorganic phosphorus was 1.3 ±0.3 mmol/l. The median values for serum ALP were 312.0 IU/l (IQR: 224.0–497.0 IU/l), serum intact PTH 4.5 pmol/l (IQR: 3.4– 10.4 pmol/l) and 25(OH)D 85.2 nmol/l (IQR 59.0–162.2 nmol/l). The mean BMD measured at 1 year after surgery at various sites was as follows: L1–L4 –0.832 ±0.149 g/cm2, FN 0.790 ±0.146 g/cm2, TH 0.822 ±0.168 g/cm2 and distal forearm 0.545 ±0.126 g/cm2. Compared to baseline, significant improvement was noticed at lumbar spine, femoral neck and total hip BMD, while change in distal forearm was not significant.
There was no long term surgical complication like vocal cord palsy or permanent hypoparathyroidism.
Discussion
In children and adolescents, PHPT is a rare condition, with literature limited to only a few case reports and retrospective analysis of hospital records. There is dearth of information on the long term consequences of the disease in children.
Primary hyperparathyroidism in children may be unique due to various reasons.
In a review of 12 studies of pediatric PHPT, Roizen et al. noted that only 15% were asymptomatic while 85% were symptomatic [24]. In our cohort, all the patients were symptomatic. The median duration of symptoms was 30 months which is comparable with a previous study from India [15].
In pediatric and juvenile PHPT, there is more extensive and severe musculoskeletal involvement. Primary hyperparathyroidism may masquerade as rickets in children and adolescents which has earlier been reported in a few case reports [4–9]. In our cohort, 50% patients presented with rachitic manifestations with genu valgum being the most predominant phenomenon. They were misdiagnosed as rickets outside our institute and were given calcium and vitamin D supplements. Hence, it is imperative for a general physician to understand that all bone deformities are not due to rickets and routine supplementation of elemental calcium should be discouraged prior to basic biochemical investigations.
Concomitant vitamin D deficiency may mask the hypercalcemia of PHPT. Many patients may actually be normocalcemic which may further lead to a misdiagnosis. In our cohort, 2 patients were normocalcemic (serum calcium < 2.6 mmol/l) with concomitant severe vitamin D deficiency. In such circumstances, PHPT should be suspected if parathyroid hormone is inappropriately elevated, not accounted by the level of vita-min D deficiency. The clinician should reassess biochemical parameters after making the patients vitamin D replete.
In the previous literature, renal disease in PHPT was found to be less severe in children, as compared to that in adults [24]. In our cohort, all the patients had normal renal functions, yet 50% had renal stones detected on ultrasonography. Previous studies from India showed a prevalence of renal stones in 30% and 40% of the patients [15, 16]. The rising trend of renal stones may be due to increased and inadvertent use of calcium and vitamin D supplements for any patient with musculoskeletal complaints.
Most children often complain of vague symptoms that may lead to a significant delay in diagnosis. In our cohort, none of the patients had any neuropsychiatric manifestation. However, 40% presented with gastrointestinal symptoms like anorexia, reflux symptoms, constipation which could have been easily overlooked.
Primary hyperparathyroidism at a younger age may represent a spectrum of genetic diseases like MEN 1 and 2A, or hyperparathyroidism-jaw tumour. Genetic analysis is warranted if there is suspicion of the same.
Two patients in our cohort had jaw tumour. The jaw lesions in PHPT may be due to brown tumour, epulis, intraoral bone swellings, bone cysts, or ossifying fibroma (hyperparathyroidism jaw tumour). One patient had significant decrease in the size of the mandibular lesion after parathyroid surgery, suggesting a possibility of brown tumour. The other patient had histologically proven ossifying fibroma, whose size did not shrink after parathyroid surgery and she underwent jaw tumour resection along with reconstruction. Hyperparathyroidism jaw tumour is a genetic disease with ossifying fibroma and parathyroid carcinoma due to mutation of HRPT2 gene encoding the protein parafibromin. Although genetic analysis was not done in our patient due to resource constraints, presence of ossifying fibroma raised a possibility of HPT-jaw tumour. Although she had a single parathyroid adenoma proven on histopathology, she is under continuous surveillance for recurrence of disease.
Pediatric PHPT is usually associated with multiglandular disease. In our cohort, all the patients had single parathyroid adenomas, diagnosed both radiologically and histologically. Despite this, there may be a high risk of recurrent disease. So, there is a need for long term surveillance.
The prevalence of hungry bone syndrome (HBS) in adults after surgery is reported to vary from from 24% to 82% in Indian studies [2, 25, 26]. Children may be at higher risk of HBS due to more prevalence of skeletal involvement. In our cohort, 40% had hungry bone syndrome, requiring aggressive management with intravenous calcium infusion in addition to oral calcium and alphacalcidol. Adequate measures should be taken to prevent HBS like vitamin D supplementation prior to surgery and early recognition of the disease before extensive skeletal involvement occurs.
In adults, there have been many long term follow-up studies following parathyroidectomy, documenting changes in various clinical, biochemical and bone densitometric parameters [25, 27–29]. However, such long term follow-up in children is lacking. We had followed-up our patients for 6 months and 1 year after surgery. Although the patients had significant clinical and biochemical improvement after one year of parathyroidectomy, yet residual bone deformities had persisted, without any further progression. A decision for correction of bone deformities was made once ALP levels had normalized.
Strengths and limitations
A major strength of our study is its prospective nature and follow-up over 6 months and 1 year. Most of the previous studies have been either case reports or retrospective and cross sectional databases.
However, a limitation is the inclusion of a smaller sample size, but that can be attributed to the rare prevalence of the disease in children.
Another limitation of this study was the lack of genetic screening of multiple endocrine neoplasia (MEN 1and 2A) and hyperparathyroid-jaw tumour (HRPT2 mutation). Although none of the patients had any syndromic manifestations of MEN1, this cannot be ruled out conclusively without genetic screening.
Follow-up beyond one year will be required to analyze the true nature of the disease in children and adolescents.
Conclusions
Primary hyperparathyroidism in children and adolescents is a rare disease that warrants early recognition. It may often masquerade as rickets leading to misdiagnosis and inappropriate management. Concomitant vitamin D deficiency may further complicate the scenario by masking the hypercalcemia. Primary hyperparathyroidism should be suspected if parathyroid hormone is unusually elevated for the vitamin D deficiency in a normocalcemic patient presenting with skeletal deformities. Although all the patients in our cohort had single adenomas on resection, they require regular surveillance to look for recurrence of disease or development of syndromic manifestations. Hungry bone syndrome is also more common in children after surgery, and this needs to be addressed early to prevent severe hypocalcemia and its complications. Eventually, there is a need for larger cohorts of pediatric and juvenile PHPT with longer follow ups to confirm our findings of clinical spectrum, outcomes of surgery, long term complications, and to be able to device an optimal follow up protocol.









