
Introduction
The term “dysmorphology” was coined by David W. Smith in 1960 to denote the medical discipline concerned with the study of congenital anomalies, with particular emphasis on the morphology of body anatomy. The term is also employed to denote individuals whose physical characteristics, whether of a specific body part or the entire body, deviate from the standards prevalent within a given societal group of similar ethnicity and age. When evaluating dysmorphia, it is essential to consider the physical features of the parents and the overall body image, not just the head or face, to avoid limiting diagnosis and misdiagnosis. The dysmorphic syndromes described in this paper are distinct, pathogenetically related groups of symptoms that include both differences in appearance and body structure, as well as organ defects arising at early stages of embryogenesis. These are rare diseases, with an incidence of no more than 1 : 2,000 births and most often less than 1 : 10,000. However, knowledge of their course, as well as diagnostic and therapeutic methods, is crucial for optimizing medical care [1, 2]. This paper addresses the issue of dysmorphic syndromes in the context of patient overgrowth syndrome, which is characterized by excessive height among children, as determined by measuring body length using an Epstein bench or infant stadiometer. Excessive growth is defined by a body length/height that is above the 97th percentile [3]. Macroglossia, omphalocele, dolichocephaly and many other features that clearly distinguish this disorder from others can also be observed in overgrowth syndrome [4]. This work is particularly noteworthy for its emphasis on the distinctions between the various disorders, a factor that is instrumental in expediting the diagnostic process and facilitating the implementation of suitable treatment [5]. All such syndromes are named after the researchers who initially described patients exhibiting specific dysmorphic features and/or contributed significantly to elucidating the genetic underpinnings.
Material and methods
A comprehensive literature search was conducted using databases such as Google Scholar, PubMed, Web of Science, Orphanet, OMIM, and the NIH database. The following search phrases were applied to ensure a thorough examination of the topic:
causes of the disease;
clinical presentation of the disease;
diagnosis of the disease;
treatment of the disease.
The selection process was guided by specific inclusion criteria to ensure the relevance, quality, and reliability of the data. Syndromes were selected based on evidence of monogenic inheritance, which served as a primary criterion for inclusion. We prioritized studies that:
Were as recent as possible, to reflect the latest scientific findings.
Included case reports, which offer detailed descriptions of clinical manifestations and treatment outcomes.
Were original, peer-reviewed, and published research articles to ensure scientific rigor.
Had full-text availability, allowing for comprehensive analysis.
Were written entirely in English, to maintain consistency and facilitate interpretation.
This methodological approach ensured a focused and genetically coherent review of overgrowth syndromes associated with dysmorphic features. Applying these criteria, a total of 955,377 studies were identified, of which 58 were included in the final analysis.
The photos included in the article were published with the consent of the patients' parents/guardians.
Results
Sotos syndrome
Sotos syndrome (OMIM: 117550, ORPHA:821) is a neurodevelopmental disorder with an estimated prevalence of 1/14,000 cases [6]. The occurrence of this disorder is determined by pathogenic variants or microdeletions in the NSD1 gene (5q35) in the majority of cases (95%), which are inherited in an autosomal dominant manner. In the rest of the cases, biallelic mutations in the APC2 gene (19p13.3) can be diagnosed [7].
The syndrome is characterized by overgrowth that commences in the antenatal and neonatal period and, in contrast to Beckwith-Wiedemann syndrome, persists throughout life, being most pronounced in childhood. At first glance, we can detect macrocephaly in such a patient, which is out of proportion to the rest of the body and persists throughout life [8]. During the neonatal period, parents can observe a range of physical characteristics and developmental issues in their offspring. These may include, but are not limited to, feeding problems, premature eruption of teeth, scoliosis, and large hands and feet. Among the more serious disorders to be considered are instances of seizure activity, conductive hearing loss, and anomalies of the cardiac or genitourinary system. It is also important to note that individuals diagnosed with the syndrome exhibit an elevated risk of developing cancer, particularly atypical and malignant forms [9]. The syndrome is characterized by a number of distinct physical facial features (Fig. 1) including a prominent face, characterized by a narrow and elongated facial structure, flushed cheeks, downward-sloping eyelid crevices, hypertelorism, a prominent forehead with a lack of hair in the frontal and temporal regions, highly arched palate and a pointed chin that becomes more prominent with age [10]. In addition to phenotypic features, patients present with excessive skin on hands and feet and hypotonia, and exhibit intellectual disabilities with speech delay of varying degrees, along with a wide spectrum of behavioral disorders [11].
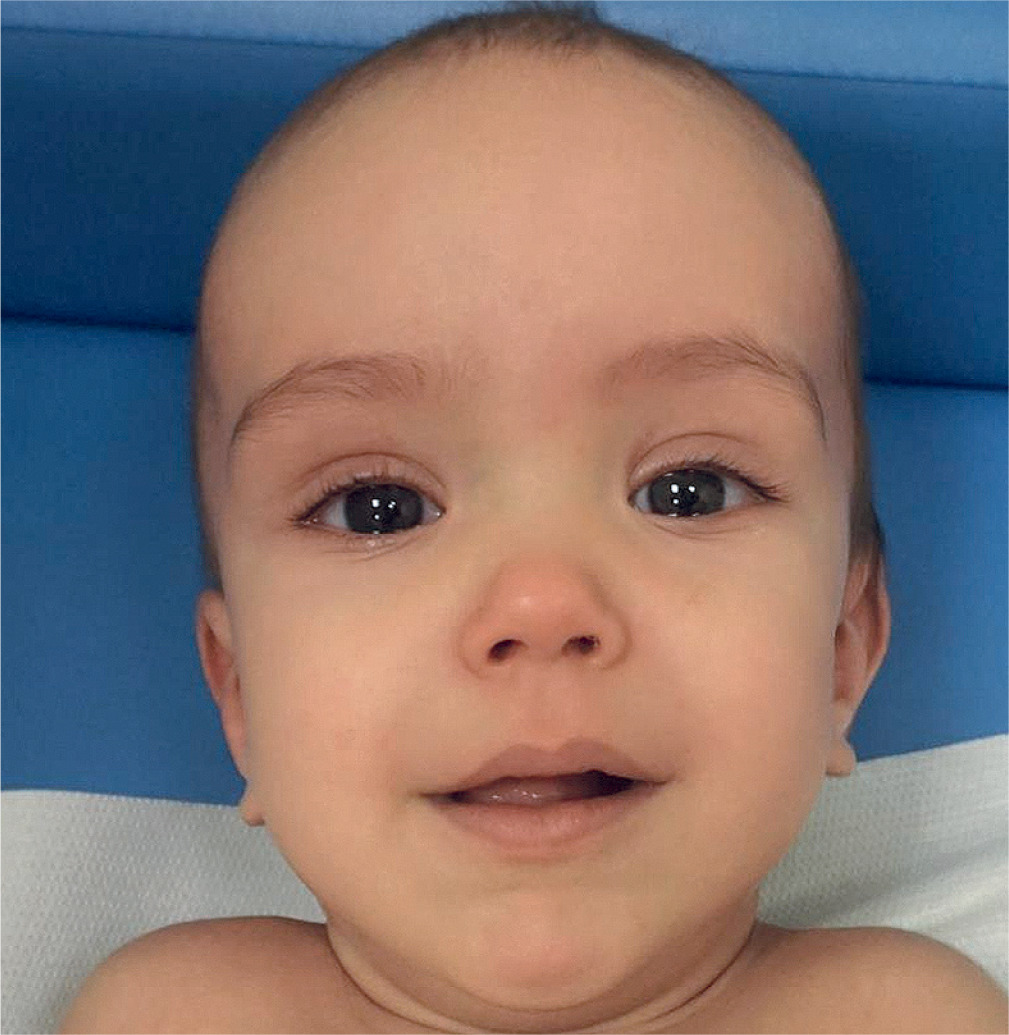
Beckwith-Wiedemann syndrome
Beckwith-Wiedemann syndrome (BWS, OMIM:130650, ORPHA:116) is a disorder with a prevalence of 1/13,700 [12, 13] characterized by excessive growth, congenital malformations and an increased predisposition to cancer. The condition arises from genetic and/or epigenetic changes, leading to dysregulation of genes located primarily on chromosomes 11p15.5 [14] and 11p15.4. Beckwith-Wiedemann syndrome occurs sporadically (85%), and familial inheritance has been observed in approximately 15% [12].
The most common feature, and the first to appear, is excessive growth, which becomes apparent as early as the second half of pregnancy (including polyhydramnios) and during the first years of life. An equally common disorder, occurring in 30–50% of newborns, is hypoglycemia, which can also be mild and transient. We can also observe the presence of macroglossia, coarse facial dysmorphic features (Fig. 2), hernias involving the anal/peritoneal/epidural/umbilical region, embryonal tumors such as Wilms’ tumor or hepatoblastoma, posterior spiral fold and anterior earlobe involvement, visceromegaly, pigmented nevi, adrenal cytomegaly and heart defects including cardiomegaly. Typically, excessive growth continues until 7–8 years of life, after which the rate slows down. It can also manifest itself through hemihyperplasia, which leads together with macroglossia to difficulties during feeding, speech and even, although rarely, sleep apnea. Growth among adults affected by this syndrome, on the other hand, does not usually indicate abnormalities.

The management of patients diagnosed with BWS involves the implementation of both medical and surgical strategies, given that, if the child survives the early childhood period, their future prognosis is generally favorable. The therapeutic approach is meticulously tailored to the individual’s presenting symptoms and their underlying causes. The treatment of hypoglycemia involves either oral feeding or glucose supplementation, while cases of hyperinsulinism are managed through standard pharmacotherapy, overseen by an endocrinologist. Surgical interventions may be necessary in cases of neoplasms, macroglossia, omphalocele and facial hemihyperplasia, and in situations where macroglossia leads to severe respiratory failure, tracheostomy and respiratory support may be necessary [15, 16].
Simpson-Golabi-Behmel syndrome
Simpson-Golabi-Behmel syndrome (SGBS, OMIM:12870, ORPHA:373) is a rare X-linked recessive overgrowth disorder that has been diagnosed in approximately 250 individuals worldwide [17]. It is caused by mutations in the GPC3 gene located on chromosome Xq26.2. [18, 19]. Female carriers of SGBS generally show milder features due to incomplete penetrance, with full phenotypic expression being an extremely rare exception to the X-linked recessive inheritance pattern [20].
The first symptoms can be observed during ultrasound examinations as early as the first trimester of pregnancy; however, they are not pathognomonic, which is why most diagnoses are made postnatally. In SGBS type I, early prenatal signs may include increased nuchal translucency (NT) detected around 12–15 weeks of gestation, although this finding is nonspecific. More characteristic features, such as fetal macrosomia (above the 97th percentile), polyhydramnios, organomegaly (enlargement of liver and spleen), renal anomalies, diaphragmatic hernia, and cardiac defects, usually become evident during the second trimester, typically after 20 weeks of pregnancy [19, 21, 22]. The spectrum of clinical symptoms is broad, primarily including tissue overgrowth and congenital defects affecting the heart, craniofacial structures, genitourinary system and skeleton. Craniofacial anomalies frequently associated with SGBS include macrocephaly, coarse facial features, hypertelorism, cleft lip and palate, lip thickening, macroglossia and a broad nasal bridge (Fig. 3) [23, 24]. Limb anomalies include fingernail hypoplasia and postaxial polydactyly. Skeletal manifestations encompass spinal deformities such as scoliosis, advanced bone age, and structural abnormalities of the ribs and vertebrae. The most frequently reported congenital heart defects include ventricular or atrial septal defects and arrhythmias. A range of genitourinary anomalies has been documented, including renal dysplasia, renal cysts, cryptorchidism, hydrocele as well as hypospadias. The disorder is additionally characterized by psychomotor impairments, manifesting as muscular hypotonia and moderate intellectual disability. Other features include congenital diaphragmatic hernia, neonatal hypoglycemia, and supernumerary nipples [25].
Initial treatment includes monitoring serum glucose levels, addressing airway obstruction and feeding difficulties resulting from congenital defects. In later stages of life, patients require neurodevelopmental and renal assessment, as well as regular full-body imaging and AFP-level testing due to higher risk of tumor development. Additional monitoring includes cardiac evaluations if needed, as well as annual ophthalmologic and hearing assessments [26].
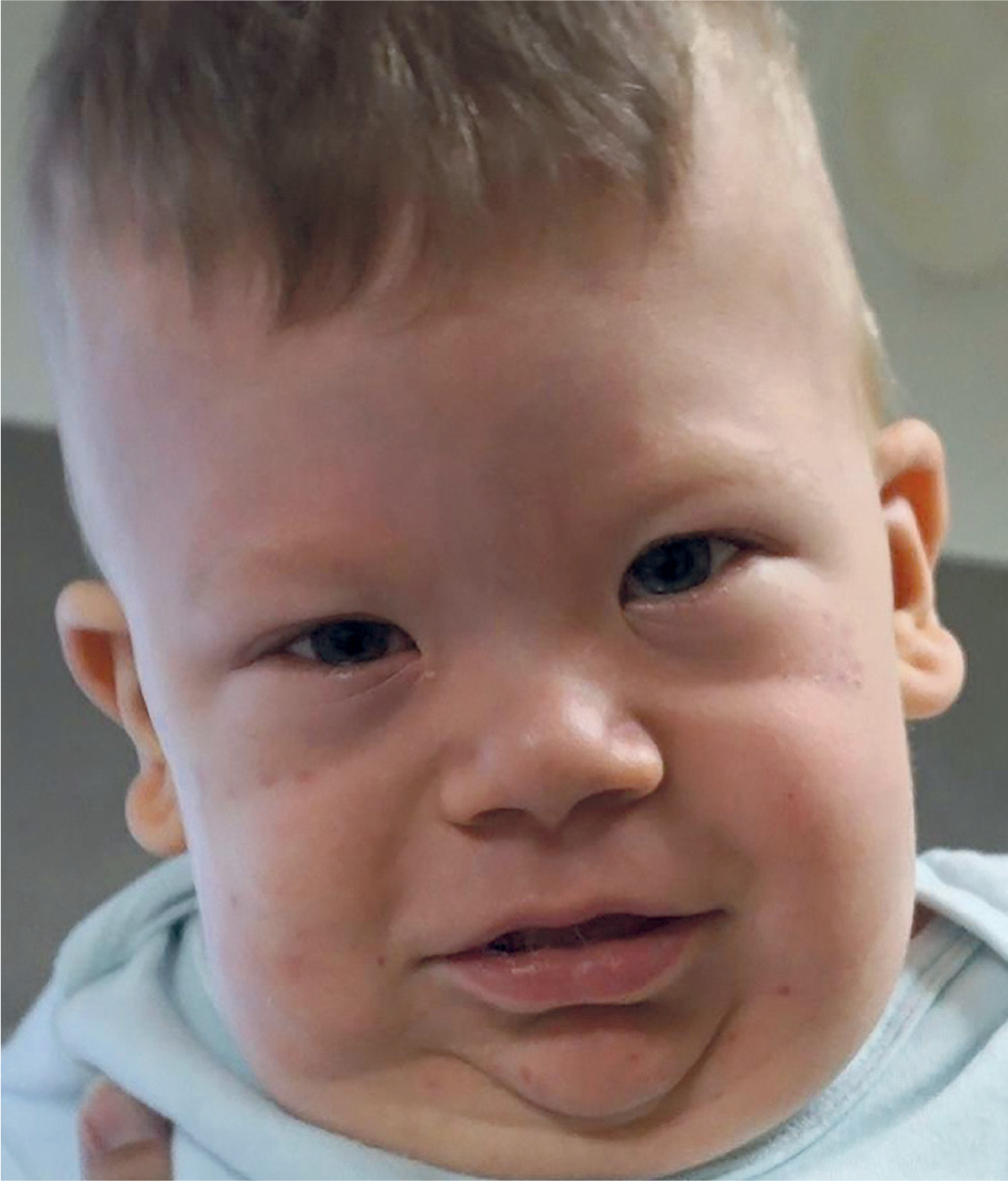
Bannayan-Riley-Ruvalcaba syndrome
Bannayan-Riley-Ruvalcaba syndrome (BRRS, OMIM:158350, ORPHA:109) syndrome is a disorder with autosomal dominant inheritance. In total, 83 pediatric cases of this disease had been reported by 2024. Most cases of BRRS are caused by mutation in PTEN, which is an antioncogene located on 10q23.31. This alteration of chromosome 10 also underlies Cowden syndrome and almost 20% cases of Proteus syndrome. These overgrowth disorders are classified as PTEN hamartoma tumor syndrome [27, 28]. Unlike Cowden syndrome, BRRS has its onset in childhood.
The syndrome is characterized by macrocephaly, intestinal polyposis, hyperpigmentation freckles on the genitalia, slowed developmental progress and dysmorphic features. In newborns, head circumference, birth weight, and birth length are above the 97th percentile; however, their growth normalizes during childhood. Adults typically reach normal body proportions. Hyperpigmentation lesions not only occur in infancy, but can also develop during adolescence [29]. Other skin abnormalities include café-au-lait macules, verrucous nodules, and subcutaneous hemangiomas compared. Hamartomatous polyps can cause anemia, abdominal pain, and blood in the stool [30]. Endocrine disorders are also common in this syndrome (Table I) [31–37]; more than half of patients develop thyroid disease [28]. Due to neurodevelopment disturbances, individuals may present intellectual disability and autism spectrum disorder. The life expectancy of patients remains unknown; however, increased risk of developing cancer may shorten their lifespan (Table II) [38].
Table I
Endocrine abnormalities potentially associated with described syndromes
Table II
Oncologic risk, tumor types, and associated life expectancy of the described syndromes
There are no approved clinical criteria to diagnose BRRS, but pigmented macules on the penis seems to be the most significant symptom of this disorder. Moreover, marked macrocephaly and gastrointestinal polyps may suggest the possibility of this syndrome. The diagnosis can be confirmed by genetic testing for PTEN mutation.
Due to PTEN mutation, a major part of BRRS management consists of regular cancer screening. Because of skin lesions, individuals should remain under dermatologist supervision [28]. Female patients should also perform breast self-examinations, because women with this mutation have a lifetime risk of breast cancer of 67–85% [39, 40].
Marshall-Smith syndrome
Marshall-Smith syndrome (MSS, OMIM:602535, ORPHA:561) is a condition with fewer than 60 cases described in the medical literature. The NFIX gene, located on chromosome 19p13, carries heterozygous mutations that are associated with MSS and are likely to have arisen de novo [41]. These mutations typically cause a dominant-negative effect, disrupting normal protein function and resulting in significant phenotypic consequences.
Characteristic symptoms include premature skeletal maturation (skeletal anomalies including progressive kyphoscoliosis, short stature), delayed development, breathing difficulties, and facial dysmorphia, which includes a high forehead, large eyes with blue, convex sclerae, a retracted chin, and a short, upturned nose. Skeletal abnormalities include uneven bone maturation and osteopenia [42]. Patients may present with umbilical hernias, excessive hair growth, connective tissue abnormalities such as joint hypermobility and excessive skin laxity, and heart abnormalities such as ventricular septal defect (VSD), atrial septal defect (ASD), aortic coarctation, pulmonary valve stenosis, mitral or aortic valve regurgitation, and hypertrophic cardiomyopathy [42]. Neurological defects are also present in this syndrome, including anomalies of the corpus callosum, ventriculomegaly and delayed myelination. Difficulties in breathing – often resulting from upper airway obstruction, laryngomalacia, or narrowing of the nostrils – pose a serious risk of death, especially among infants [43]. Developmental delay can be divided into moderate or severe and is often accompanied by limited speech and intellectual disability. In addition, behavioral abnormalities such as strong attachment to objects such as toys are often observed. Those who survive into later stages of life commonly experience serious intellectual disabilities, limited speech abilities, and skeletal issues, including kyphoscoliosis and osteopenia [44].
Treatment of MSS requires a multifaceted approach, emphasizing respiratory support, dietary solutions and prevention of skeletal complications. In many cases, upper airway obstruction can be relieved with tracheostomy or nasopharyngeal airway support, and early treatment can significantly improve survival rates. Bisphosphonate treatment is also used to treat osteopenia and reduce the fracture risk in some patients [42]. Regular monitoring for problems such as pulmonary hypertension, cervical spine stenosis, and developmental delays is also crucial.
Weaver syndrome
Weaver syndrome (WVS, OMIM:277590, ORPHA:3447) is an uncommon hereditary disorder; 50 cases have been reported in the medical literature so far. It is characterized by prenatal and postnatal overgrowth distinctive craniofacial features, and advanced bone age; in particular, the development of the carpal bones is advanced relative to that of the rest of the hand [24]. Weaver syndrome is essentially caused by transformations within the EZH2 quality, found on chromosome 7q36 [45]. This epigenetic dysregulation leads to abnormal growth and development [45, 46]. About half of cases are due to de novo mutations, while the other half are inherited in an autosomal dominant manner. However, some individuals may carry the mutation without apparent symptoms, complicating genetic counseling and risk assessment [45, 46].
In some cases, WVS may be associated with mutations in other genes, such as EED (currently referred to as Cohen-Gibson syndrome) or SUZ12, which also affect epigenetic regulation. These mutations can lead to similar symptoms, making differential diagnosis difficult [47].
The hallmark of WVS is excessive growth, often visible before birth. Prenatal ultrasound can reveal macrosomia (larger than average size), and newborns tend to have greater birth weight and length. Snoring or low-pitched crying is a common early symptom [45, 46]. Craniofacial features include a broad forehead, widely spaced eyes (hypertelorism) [46, 48], large or low-set ears, a long philtrum, and micrognathia (a small lower jaw) (Fig. 4). These features become more pronounced during early childhood [45, 46]. Other skeletal abnormalities include advanced bone age, pamprodactyly (permanently bent fingers), and joint contractures. Some patients may also exhibit scoliosis or kyphosis [45, 48]. Some patients also experience bone demineralization, especially in the hands and feet, which can lead to increased susceptibility to fractures. Developmental delays and intellectual disabilities are common and can range in severity from mild to profound. Hypotonia or decreased muscle tone along with poor coordination is often observed. These challenges often affect motor skills and daily functioning [45, 46].
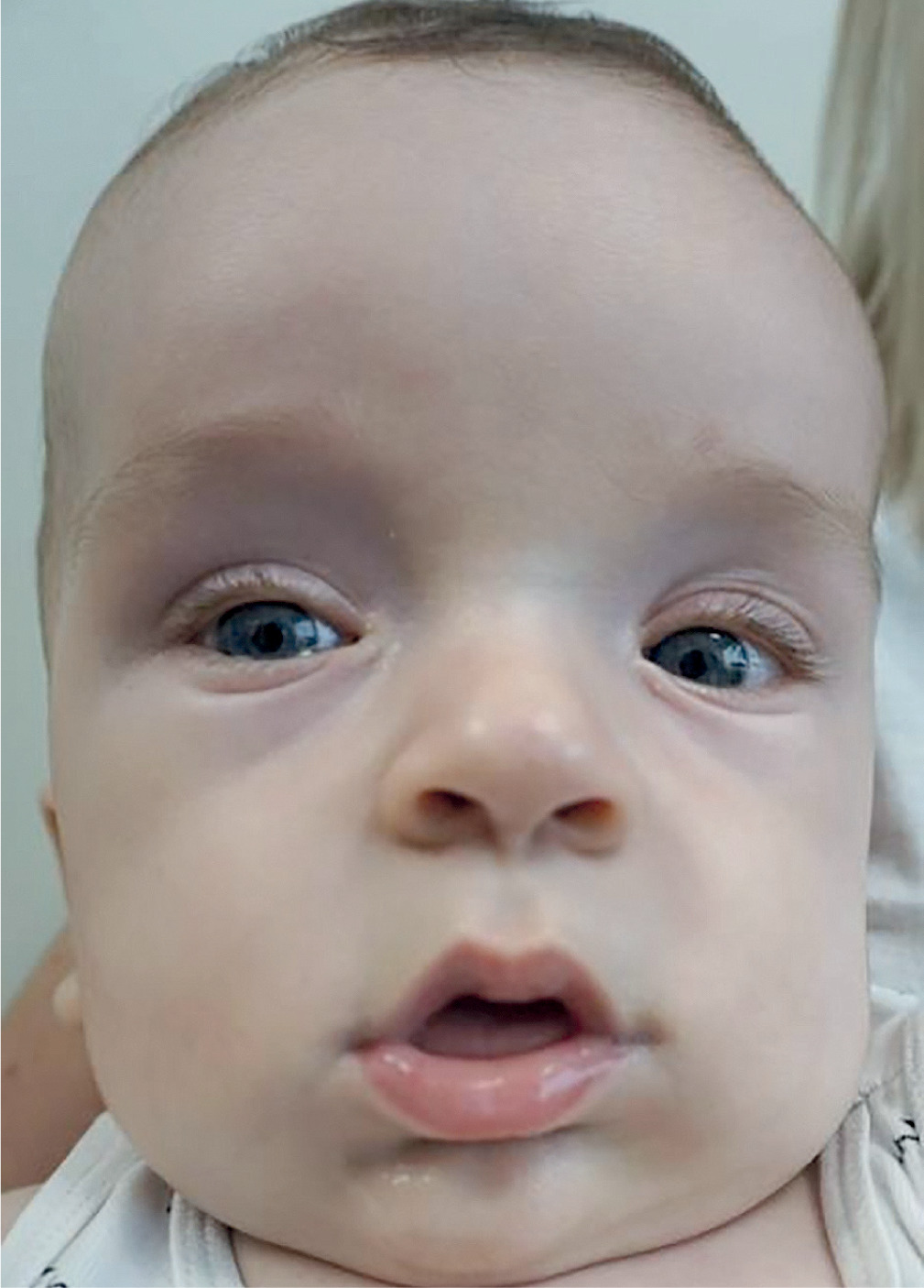
Elejalde syndrome
Elejalde syndrome (OMIM:200995, ORPHA:221054), also known as acrocephalopolydactylous dysplasia, is a lethal disorder inherited in an autosomal recessive manner. Due to the very limited number of reported cases (fewer than 10), the causative gene has not yet been identified [49]. It has been hypothesized that the syndrome may be associated with excessive proliferation of fibroblasts. Affected individuals typically present with a swollen globular body, thickened skin, and short neck with redundant folds. Craniosynostosis is frequently observed, along with increased birth weight and macrocephaly. Distinct craniofacial features include hypertelorism and epicanthic folds, as well as hypoplastic nasal bones. Additional findings may include hypoplastic lungs and omphalocele. Visceral organ involvement is common, with organomegaly such as hepatomegaly, nephromegaly and cystic kidneys. Limb anomalies such as polydactyly and micromelia are also reported [50].
Nevo syndrome
Nevo syndrome (OMIM:225400, ORPHA:2691) has been identified as an allelic form of the kyphoscoliosis type VI Ehlers-Danlos syndrome (EDSKSCL1), which is characterized by a homozygous or compound heterozygous mutation in the gene encoding lysyl hydroxylase, located on chromosome 1p36 [51, 52]. It is inherited in an autosomal recessive manner, and to date, approximately seven cases of patients with this syndrome have been described.
Patients diagnosed with Nevo syndrome typically exhibit a combination of distinctive physical characteristics. These include elevated height, delayed motor development, and progressive kyphoscoliosis that progresses rapidly. It is frequently observed that newborns present with hypotonia, atypical contractures of the upper and lower limbs, scoliosis, and torticollis. The presence of joint hypermobility can manifest as an unstable gait, while the skin, characterized by its soft and velvety texture, is predisposed to bruising and scarring following minor injuries. Additionally, patients may present with abnormalities such as spina bifida at the L5-S1 level, bilateral inguinal hernias, spindle-shaped fingers, and wrist drooping [53].
The diagnosis of the syndrome can be made on the basis of the patient’s clinical picture and the careful observation of the parents during the first years of the child’s life. The presence of increased body length, wrist droop, muscular hypotonia, and kyphoscoliosis at birth should prompt the doctor to consider a diagnosis of Nevo syndrome [53, 54]. Patients with an initial diagnosis should then undergo a detailed biochemical analysis. The course of the study may reveal elevated urinary lysyl-pyridine/hydroxylysine ratios [54, 55], hydroxylysine deficiency in hydrolyzed dermis analysis, and reduced enzyme activity in cultured skin fibroblasts [56, 57].
High doses of ascorbic acid were used during treatment, resulting in substantial enhancements in muscle strength and accelerated wound healing. Consequently, more intensive treatment with vitamin C enabled the body’s repair processes to be optimized, culminating in enhanced muscle function and a reduced recovery time following injury [55].
Tatton-Brown-Rahman syndrome
Tatton-Brown-Rahman syndrome (TBRS) (OMIM: 615879, ORPHA: 404443) is a genetic disorder characterised by postnatal overgrowth and developmental delay. To date, approximately 100 cases of TBRS have been reported. The condition is caused by pathogenic variants in the DNMT3A gene, which in most cases arise de novo [58, 59].
Information regarding birth weight and length in newborns with TBRS is limited. While not all affected infants are macrosomic, most individuals develop features of overgrowth later in life, including tall stature and macrocephaly. During early infancy, hypotonia and seizures may occur. Developmental delay is usually evident from early childhood and can range from mild to severe, though it most often remains moderate. Facial dysmorphic features typically become more apparent during adolescence and may include short palpebral fissures, a round face, horizontally thick and low-set eyebrows, and enlarged central incisors. Additional clinical findings may include kyphoscoliosis, joint hypermobility, behavioural problems, cardiovascular abnormalities, cryptorchidism and an increased risk of malignancy [59–61].
Initial diagnosis is based on clinical evaluation, with the most characteristic features being generalised overgrowth and developmental delay or intellectual disability. The diagnosis can be confirmed through molecular genetic testing, which demonstrates the presence of a heterozygous pathogenic variant in the DNMT3A gene.
While most patients do not require specific treatment, they do benefit from education about potential complications and regular monitoring of symptoms for both themselves and their families. Physical therapy is recommended to enhance mobility and prevent orthopaedic complications. Those with hypertonia or dystonia may require pharmacological management with agents such as baclofen, tizanidine, Botox®, or anti-Parkinsonian medications [59, 60].
Conclusions
Overgrowth syndromes are rare genetic disorders that require comprehensive diagnostic evaluations and multidisciplinary treatment strategies. Diagnosis is primarily based on clinical assessment of characteristic features, such as overgrowth patterns and dysmorphic features. Genetic testing plays a key role in confirming the diagnosis by identifying mutations (next generation sequencing technique) or epigenetic alterations in specific genes (MS-MLPA method). In some cases, symptoms may be detected during prenatal ultrasound examinations; however, the vast majority of diagnoses are made after birth.
There is no causal treatment for these syndromes. Management focuses on multidisciplinary care tailored to the individual needs of each patient and varies depending on the specific case. It may include surgical correction of congenital anomalies, pharmacological treatment for metabolic disorders or seizures, and regular monitoring for tumor development due to the increased risk of malignancies associated with many of these syndromes. Tumor surveillance typically involves routine imaging studies aimed at early detection of neoplasms. Developmental support, including speech therapy, specialized education, and physiotherapy, is also crucial, particularly in patients with intellectual disabilities or musculoskeletal abnormalities. Lifelong monitoring and personalized treatment plans are essential for improving long-term outcomes and enhancing the quality of life for affected individuals.









