
Introduction
Neonatal severe primary hyperparathyroidism (NSHPT) is a rare autosomal recessive calcium homeostasis disorder that occurs shortly after birth, characterized by striking hyperparathyroidism, marked hypercalcaemia, and hyperparathyroid bone disease [1]. Hypotonia, respiratory distress, bone fractures, intestinal dysmotility, and growth retardation are seen in patients with NSHPT, and their neurological development can be significantly affected. NSHPT can be fatal if left untreated [1]. NSHPT is caused by a loss of function of the calcium-sensing receptor (CASR), whose gene is encoded in the long arm of chromosome 3 (3p-13.3- 21) [2]. The extracellular calcium-sensing receptor (CASR) is from a family of G-protein-coupled receptors (GPCR) that are expressed in many locations, including parathyroids and kidneys. It plays a key role in maintaining extracellular calcium balance. To date, many different diseases caused by mutations of CASR have been reported [2]. While gain-of-function mutations result in hypokalaemic disorders of autosomal dominant hypocalcaemia and Bartter syndrome type V, loss of function mutations cause 3 hypercalcaemic disorders: familial hypocalciuric hypercalcaemia (FHH), neonatal severe hyperparathyroidism, and primary hyperparathyroidism [3]. Herein we report on a newborn male infant with NSHPT caused by previously reported homozygous CASR gene mutation. Although this mutation had been reported unresponsive to cinacalcet therapy, the patient’s normocalcaemic state could be maintained by cinacalcet with a calcium-restricted diet until the age of 13 months.
Case report
A 6-day-old boy was referred to our clinic with a high calcium level of 23.7 mg/dl from a distinct hospital where he presented with the complaint of poor feeding. There was no problem in the prenatal follow-up of the patient, who was born at full term, weighing 3600 g (0.45 SD), by normal vaginal route. Vitamin D prophylaxis had not been initiated in the patient, who was fed exclusively with breast milk. The patient, whose parents were third-degree relatives, had 4 healthy siblings (3 girls, 1 boy). His paternal aunt had a history of parathyroidectomy at the age of 38 years, but the reason was not known (Fig. 2). On physical examination, his weight, height, and head circumference were 3600 g (10th–25th percentile), 54 cm (50th–75th percentile), and 34 cm (10th–25th percentile), respectively.
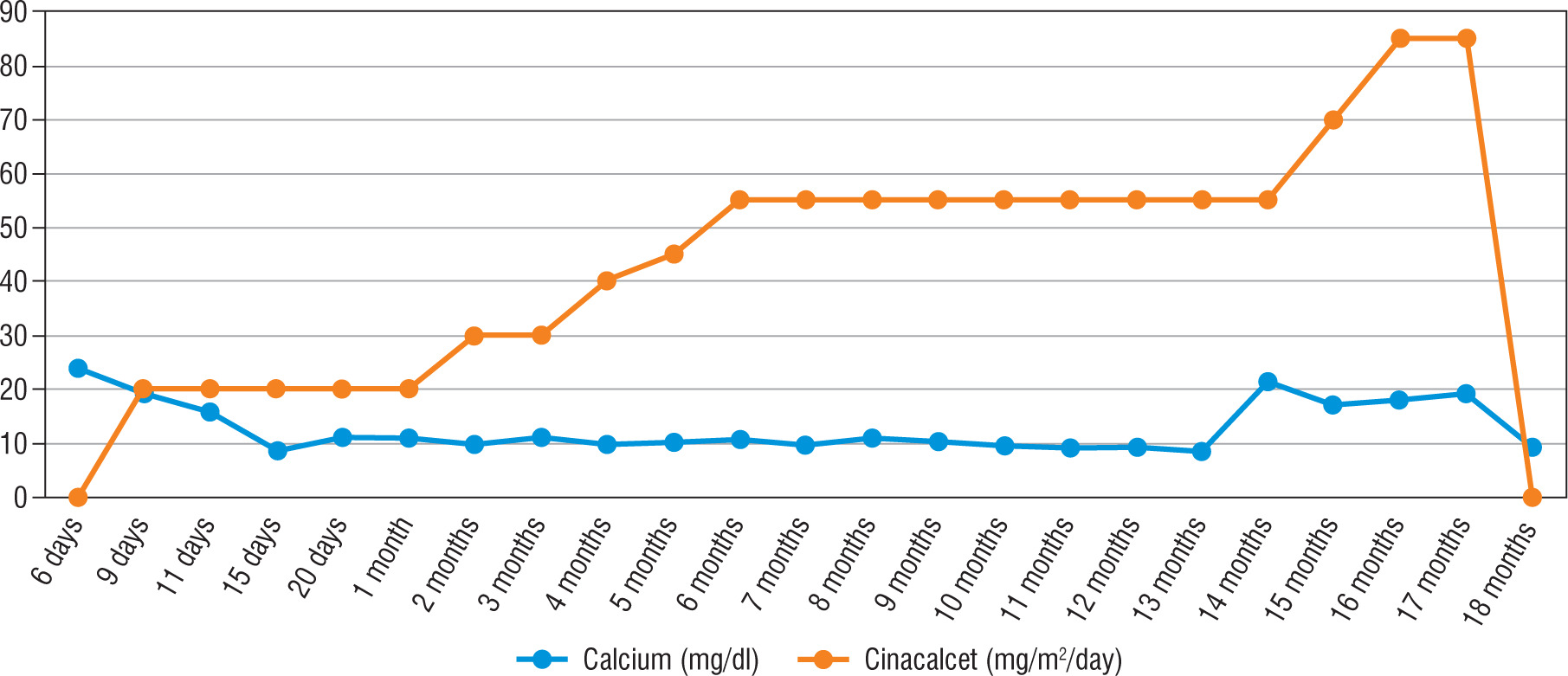
His heart rate was 134 bpm, with a respiratory rate of 134/min, blood pressure of 84/47 mmHg, and body temperature of 36.9°C. The patient was noted to have sunken fontanelle, severely decreased skin turgor, moderate hypotonia, and absent primitive reflexes. He had no dysmorphism features. Examination of other systems was unremarkable. Laboratory tests were as follows, serum calcium, 23.7 mg/dl (reference range, 9–11 mg/dl); phosphorus, 3.7 mg/dl; magnesium, 2.9 mg/dl; alkaline phosphatase, 216 IU/l; parathyroid hormone (PTH), 1120 ng/dl (reference range, 11–88 ng/dl); urinary calcium/creatinine, 0.04 (< 0.8); 25-hydroxyvitamin D, 8.57 ng/dl. Complete blood count, sodium, potassium, chloride values, liver functions, and creatinine values were normal. Serum urea level was slightly higher at 63 mg/dl (reference range, 9–48 mg/dl). Electrocardiography showed a shortening of the QT interval (300 milliseconds) and a change in the ST segment. There was no demineralization or fracture found on bone X-ray (Fig. 1). The diagnosis of severe hyperparathyroidism of the newborn was considered with clinical and laboratory (elevated serum Ca and PTH) findings, and genetic analysis studies were initiated. Together with hydration, the patient received pamidronate 2 times in a total of 1 mg/kg/dose within 48 hours. Control calcium value (19.2 mg/dl) was unresponsive to this treatment. Thereupon, cinacalcet at a dose of 20 mg/m2/day and a calcium-poor formula (basic-CaD [35mg Ca/100 g]) were started on the 3rd day of the follow-up. Serum Ca (9.7 mg/dL) returned to normal on the 6th day of treatment. After 2 weeks, the patient was discharged with cinacalcet therapy at 20 mg/m2/day, vitamin D 400 UI/day, and calcium-restricted formula. The calcium value at the time of discharge was normal (10.3 mg/dL) (Fig. 4). The patient was followed up with monthly controls. His nutrition, cinacalcet, and vitamin D doses were adjusted to ensure normocalcaemia. When he was one year old, his neuromotor development was compatible with his age. Millimetre-sized echogenicity developed in the medullary pyramids of both kidneys (nephrocalcinosis). No side effects such as vomiting or diarrhoea were observed in the patient during cinacalcet therapy. In genetic analysis, a homozygous NM_001178065.2(CASR):c.222_226delGATAT (p.Met74IlefsTer24) frameshift deletion was defined. According to American College of Medical Genetics and Genomics (ACMG) 2015 classification [4], this frameshift deletion was classified as “likely pathogenic” due to the previous reports confirming that null variants of CASR is the mechanism of disease (PVS1 according to ACMG 2015 guidelines) [5]. Another homozygous variation, NM_000388.4(CASR):c.740C>T, predicted to cause p.Ser247Phe missense amino acid change in CASR if expressed, was defined in the proband, but this was a variant of unknown significance according to the ACMG 2015 guidelines and previous reports [6]. Other family members were found as heterozygous carriers (Fig. 3). In the 13-month physical examination, body weight was 8350 g (3rd–10th percentile) and height was 72 cm (3rd–10th percentile). All system examinations were normal. Thereupon, milk and dairy products were added to his diet with a calcium amount of 250 mg/day and daily 400 UI vitamin D. One week later, his serum calcium level increased to 9.8 mg/dl and to 21.3 mg/dl in the 2nd week. Cinacalcet dosage was gradually increased to 85mg/m2/day. Hypercalcaemia was not treated effectively despite increased doses of cinacalcet. Therefore, total parathyroidectomy was carried out without autotransplantation in the patient at 17 months of age. In current histopathological evaluation, the parathyroid glands are slightly larger than normal size and poor in fat. Clear cells are evident, but there are also essential cells. The presence of these findings in all 4 glands is consistent with NSHPT. He is maintaining a diet with normal calcium, phosphate, and alkaline phosphatase with calcitriol (0.05 µg/kg/day) and calcium supplements (75 mg/kg/day). He has achieved appropriate neuro-motor milestones according to at his age of 2 years.
Figure 2
Pedigree of the patient showing affected and unaffected family members. Genetic testing was performed only on the first-degree relatives (parent, brother, sister) of the patient.Parents and siblings were found to be carriers. The aunt has a suspected history of parathyroidectomy
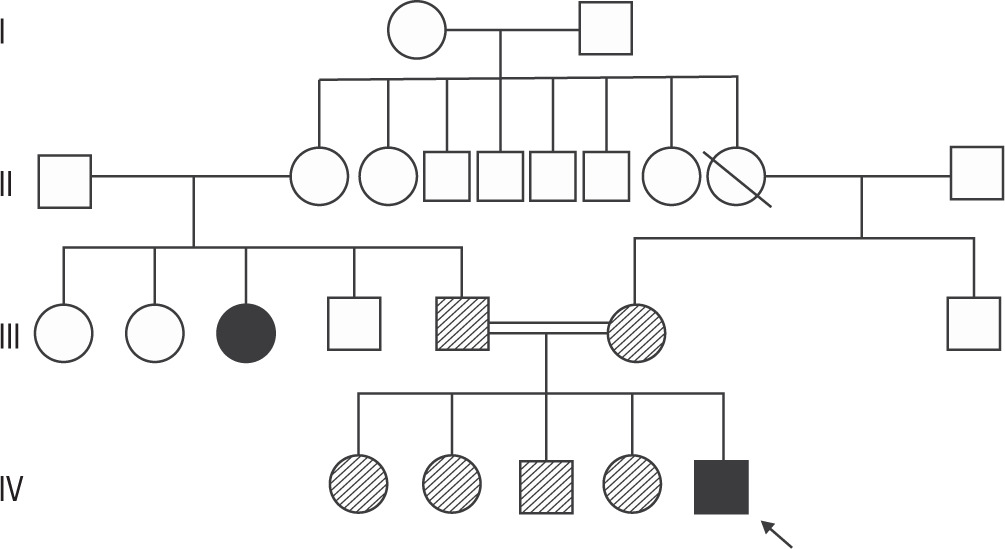
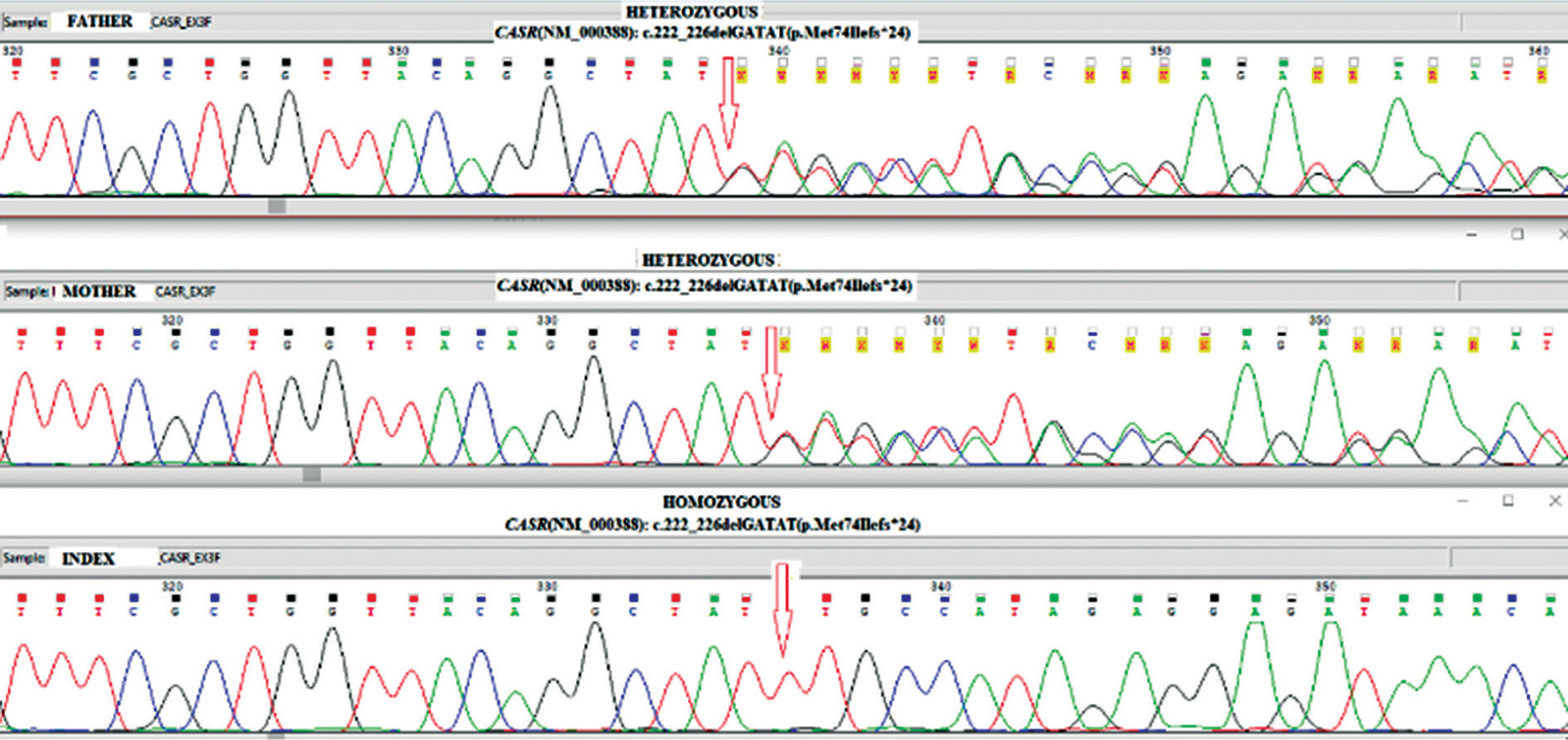
Figure 3
Identifying CASR alterations in family members. Nucleotide sequence of part of CASR after PCR amplification of genomic DNA followed by direct analysis to show the sequence of both alleles. The proband is homozygous and both parents are heterozygous for a c.222_226delGATATmutation
Molecular analysis
The entire coding region of the CASR gene was sequenced using the next generation sequencing method. For this purpose, we designed primers to amplify the coding region of the CASR gene via long-range polymerase chain reaction. The amplicons were then pooled and purified using the Agencourt AMPure XP system (Beckman Coulter, Pasadena, CA, USA). The starting DNA library was quantified using the Qubit dsDNA BR Assay kit (Invitrogen, Carlsbad, CA, USA). Sequencing library construction was performed by Nextera XT kit (Illumina, San Diego, CA, USA), which uses transposon to fragment the ends and simultaneously adds adapter and barcoding sequences. The pooled and barcoded libraries were subsequently sequenced on the MiSeq sequencer (Illumina Inc., San Diego, CA, USA).
Analysis of the NGS reads were done using IGV 2.4.8 (www.broadinstitute.com). We confirmed the mutation, and segregation analyses were done via the gold standard method of Sanger Sequencing using internal primers targeting the mutation region. The Big Dye Terminator Kit V3 was used for dideoxy sequencing, and products were electrophoretically separated on ABI 3730 XL.
Discussion
NSHPT is a rare autosomal recessive inherited life-threatening disease characterized by marked hypercalcaemia, hyperparathyroidism caused by diffuse parathyroid hyperplasia, and skeletal demineralization due to biallelic inactivation of the CaSR gene (homozygous or compound heterozygous mutations) [7].
Hypercalcaemia in NSHPT has a multifactorial aetiology and is not simply secondary to extensive bone resorption. In NSHPT, uncontrolled hyperparathyroidism is thought to stimulate osteoclasts and lead to a significant increase in bone resorption, exacerbating hypercalcaemia [7]. Medical management of NSHPT is often difficult and complex. Hypercalcaemia treatment is usually initiated using hydration, a loop diuretic, bisphosphonates, and calcitonin administration. Additionally, calcium intake must be restricted. Calcitonin may decrease bone and renal tubular calcium absorption, but its effect has been described as transient [8]. Bisphosphonates are used in children for hypercalcaemia, which inhibit osteoclastic bone resorption. Pamidronate has limited efficacy and does not specifically address the problem of abnormal CASR function [7]. Our patient was unresponsive to hydration and pamidronate treatment.
Calcimimetics are drugs that interact with the transmembrane protein of CASR, making the receptor more sensitive to calcium. Calcimimetics suppress PTH levels and increase renal Ca excretion to provide normocalcaemia, thus avoiding the need for surgery. Cinacalcet, a type II calcimimetic, is a positive allosteric activator of CASR [8]. Cinacalcet is the only drug approved for use in the medical treatment of hyperparathyroidism and parathyroid carcinoma in adults [9]. Although the safety and efficacy of cinacalcet has not yet been approved by the FDA in the paediatric age group, it has been used for life-saving purposes in many cases of FHH and NSHPT after obtaining informed consent [1, 7, 9-21] (Table I). Although we found a homozygous mutation in our patient that was previously reported to be unresponsive to cinacalcet [7], we were able to achieve normocalcaemia with cinacalcet and a calcium-restricted diet until 13 months of age. During the 13-month follow-up of the patient, calcium was added to the diet because of borderline hypocalcaemia. Later, the patient developed hypercalcaemia, and although we gradually increased the dose of cinacalcet to 85 mg/m2/day, the hypercalcaemic state could not resolved.
Table I
Published cases with neonatal severe hyperparathyroidism treated with cinacalcet
| Ref. | Age, sex, ethnicity | Ca (9–11 mg/dl) | p (4.5–6.7 mg/dl) | mg (1.6–2.3 mg/dl) | PTH (11–88 pg/ml) | Clinical findings | Age of cinacalcet, treatment response, dose | Mutation | Last visit age | |
|---|---|---|---|---|---|---|---|---|---|---|
| Abdullayev et al. [22] | 7 days Male Turkish | 17.9 | 1.7 | 1.4 | 1835 | Weight loss,jaundice | ? | p.G613E (c.1836 G>A) homozygous | 20%(n = 4) | |
| Ahmad et al. [15] | 60 days Female Arabic | 22.4 | 2.8 | 2356 | Failure to thrive | 2 months 4 mg/kg/d unresponsive? | VS4-19 (c.1378-2A>G) homozygous | 18 months Parathyroidectomyat 3rd month | ||
| Atay et al [7] | 21 days Female Turkish | 19 | 2.5 | 1.6 | 1096 | Pneumonia,respiratory distress | 28 days 30 mg/m2/d up to 90 mg/m2/d unresponsive | p.M74Ifs*24 (c.222_226del GATAT) homozygous | 14 months Parathyroidectomyat 45th day | |
| Capozza et al. [17] | 8 days Female Italian | 28.7 | 2 | 934 | Poor feeding,lethargy, cardiac arrest | 18 days 0.4 mg/kg/d up to 4 mg/kg/d unresponsive | IVS5+1G > A c.1608+1G>A Splice site-skipped exon 5 homozygous | 77 days Parathyroidectomyat 37th day | ||
| Fisher et al. No. 1 [12] | 11 months Male American | 12.9 | 4.4 | 3.1 | 76 | Jeune’s syndrome | 13 months 3.7 mg/m2/d up to 90 mg/m2/d responsive | p.R185Q (c.554G>A) heterozygous- de novo dominant negative | 32 months on cinacalcet | |
| Fisher et al. No. 2 [12] | 26 days Male American | 13.3 | 4.4 | 2.4 | 196 | Failure growth cough | 4 months 2 mg/kg/d responsive | p.R185Q (c.554G>A) heterozygous- de novo dominant negative | 13 months on cinacalcet | |
| Forman et al. [18] | 3 days Male American | 13.2 | 3.5 | N | 934 | Oxygen desaturation with feeding | 7 days 0.4 mg/kg/d up to 5 mg/kg/d (112.5 mg/m2/d) Responsive | p.R185Q (c.554G>A) heterozygous- de novo dominant negative | 14 months on cinacalcet | |
| Gannon et al. [11] | 2 days Male American | 12.9 | 2.9 | 2.7 | 1154 | Hypotonia, lethargy, apnoea | 15 days 6 mg/m2/d (responsive) up to 202 mg/m2/d | R185Q (c.554G>A) heterozygous- paternal dominant negative | 18 months cinacalcet | |
| Garcia et al. [11] | 23 days Female Spanish | 23.1 | 1.3 | 518 | Failure to thrive, hypotonia | 60 days (unresponsive)10 mg/m2/d up to 25 ng/m2/d | p.R465Lfs*9 (c.1392_ 1404del13) homozygous | 17 months Parathyroidectomyat 4th month | ||
| Hashim et al. [19] | 10 days Ceylonese Male | 29.7 | 2.3 | 403 | Fever, poor feeding, constipation | 6 weeks 1.5 mg/kg/d up to 11 mg/kg/d unresponsive | p. R227X (c.679 C>T) homozygous | 18 months parathyroidectomyat 3rd month | ||
| Kersin et al.[20] | 7 days Male Turkish | 17.9 | 1.7 | 1.4 | 1835 | Respiratory distress | 2 days responsive | p.G613E (c.1836 G>A) homozygous | 18 months on cinacalcet | |
| Murphy et al. [14] | 4 days American | 36.8 | 3.2 | 3.4 | 868 | Poor feeding, weight loss, hyperbilirubinemia, hypoxia, hypercapnea | 18 days 6 mg/m2/d up to 143 mg/m2/d unresponsive | p.R69H (c.206G>A) homozygous | 18 months parathyroidectomyat 50th day | |
| Sun et al. [21] | 7 days female Chinese | 24 | 1.7 | 4.5 | 945 | Poor feeding, constipation, pneumonia | 72 days 30 mg/m2/d up to 45 mg/m2/d responsive | p.I81K (c.242T>A) homozygous | 10 months on cinacalcet | |
| Savaş erdeve et al. [16] | 12 days Male Türkish | 19.4 | 3.3 | 2536 | Constipation restlessness | 29 days 10 mg/m2/d up to 25 mg/m2/d unresponsive | p.R544* (c.1630C>T) homozygous | 18 months parathyroidectomyat 70th | ||
| Reh et al. [13] | 7 days Female Hispanic | 12.9 | 4.8 | 2.4 | 663 | Failure to thrive, hyperbilirubinemia | 24 days 20 mg/m2/d | R185Q (c.554>A) Heterozygous de novo Dominant negative | 17 months on cinacalet | |
| Our case | 6 days Male Türkish | 23.7 | 3.7 | 2.9 | 1120 | Failure to thrive, dehydration hypotonia | 9 days 20 mg/m2/d up to85 mg/m2/d | p.Met74Ilefs * 24 homozygous | 24 months parathyroidectomyat 17 months old | |
NSHPT has been identified in children born to consanguineous FHH parents, and thus is considered to be the homozygous phenotype of FHH. Indeed, NSHPT occurring in the offspring of consanguineous FHH families or unrelated individuals with FHH has been shown to be due to homozygous or compound heterozygous CASR mutations, respectively [13]. Thus, loss-of-function CASR mutations have a gene dosage effect, with an abnormality in a single CASR allele leading to the mildly hypercalcaemic disorder of FHH, while mutations of both alleles result in NSHPT. However, individuals with sporadic NSHPT have been reported in association with de novo heterozygous CASR mutations [14]. The parents of our patient were third-degree relatives. Although heterozygous mutation was found in the parents and siblings, no family member was diagnosed with FHH. On the other hand, his paternal aunt underwent parathyroidectomy with an unknown reason. She might be diagnosed as having FHH.
The type of mutation may affect the severity of the clinical symptom and response to treatment. Cinacalcet response has also been associated with the CASR genotype in NSHPT cases. For this reason, it has been suggested that the type of mutation affecting the CASR be rapidly evaluated to determine whether a calcimimetic treatment is appropriate [1]. Various CASR mutations with cinacalcet treatment failure have also been reported, as shown in Table I. As in our case, the importance of quickly determining whether there is a place for a calcimimetic in the clinical management of a particular patient is emphasized. This can be achieved by an empirical trial of the drug as monotherapy and, if possible, a rapid assessment of the CASR genotype [7]. In our patient, we started empirically cinacalcet treatment before the genetic analysis was completed, and we were able to achieve normocalcaemia with this treatment. Even though we found a mutation that was reported to be unresponsive to cinacalcet in the genetic analysis of our patient, we continued the treatment because of the response we received. Unlike the other patient with the same mutation [7], the fact that we had a partial response to cinacalcet treatment in our patient at 13 months until calcium was added to the diet suggests that factors other than genotype may also be effective in determining the response.
In conclusion, the rapid and persistent clinical and biochemical response to cinacalcet in patients with NSHPT justifies the consideration of a calcimimetic therapy trial in patients with NSHPT to avoid parathyroidectomy and minimize the need for repeated IV bisphosphonate administration. However, parathyroidectomy is a complex procedure, and many medical centres do not have experience with parathyroidectomy in infants. We think that cinacalcet therapy can always be tried in patients with NSHPT, regardless of the type of mutation, to determine its effect in delaying parathyroidectomy, even if there is no sustained response. Thus, the surgical treatment of the patient could be delayed a little further with cinacalcet therapy.









